Células humanas são constantemente expostas a uma variedade de DNA danificando agentes de várias origens. Fontes exógenas consistem principalmente em exposição a radiações, produtos químicos (incluindo agentes quimioterápicos e alguns antibióticos), e vírus, enquanto as principais fontes endógenas incluem erros na replicação do DNA e estresse oxidativo. Os efeitos diretos da exposição genotoxica podem variar de uma base modificada a uma quebra de duplo fio de DNA potencialmente letal (DSB), dependendo do estresse e da dose de exposição. Em última análise, danos de DNA não reparados ou mal reparados podem levar ao acúmulo de mutações, rearranjos genômicos, instabilidade do genoma e eventualmente levar à carcinogênese1. As células mamíferas desenvolveram caminhos complexos para reconhecer tipos específicos de dano de DNA2,3 e repará-los em tempo hábil, sincronizados com a progressão do ciclo celular.
A radiação ionizante (IR) danifica a dupla hélice do DNA e cria quebras de dois fios (DSBs), uma das formas mais deletérias de dano de DNA. O complexo MRN (MRE11, RAD50, NBS1) funciona como um sensor de extremidades de DNA e ativa a proteína quinase ataxia telangiectasia mutada (ATM)4,5. Após a ativação inicial do ATM por extremidades de DNA, o ATM desencadeia uma cascata de eventos de DDR no local do rompimento, iniciando com um evento-chave, a fosforilação da variante histona H2AX6. Fosforilação H2AX sobre resíduo S139 ativa-o em γH2AX, abrangendo regiões até megabases em torno da lesão de DNA6,,7,,8,9. Este evento aumenta a acessibilidade ao DNA, levando ao recrutamento e acúmulo de outras proteínas de reparação de DNA7. Como o γH2AX é abundante e especificamente induzido em DSBs circundantes, ele pode ser facilmente visualizado usando anticorpos específicos, e é comumente usado como um marcador substituto para DSBs no campo de reparação de DNA. Uma vez que a ruptura é sinalizada, as células ativam suas vias de reparação de DNA e processam o dano de DNA. A proteína MDC1 (mediadora da proteína de ponto de verificação de dano de DNA 1) liga diretamente γH2AX10,interage com o ATM11 e também com NBS112,13. Contribui para aumentar a concentração do complexo de MRN no DSB e iniciar um ciclo positivo de feedback atm. γH2AX é rapidamente removido uma vez que a ruptura é reparada, consequentemente, permitindo o monitoramento da liberação DSB. Seguida pela microscopia, a diminuição do γH2AX ao longo do tempo fornece uma medição indireta de quebras residuais e eficiência de reparação de DNA.
As células eucarióticas podem reparar DSBs por várias vias, sendo as duas principais não homólogos a junção final (NHEJ) e recombinação homólogo (HR)(Figura 1). NHEJ essencialmente liga extremidades de dupla cadeia de DNA sem o uso de homologia estendida e opera durante todo o ciclo celular14,15. O RH torna-se predominante durante as fases S e G2, e é reprimido de outra forma, uma vez que requer uma cromátide irmã como modelo homólogo para reparo14,16. A escolha do caminho entre nhej e RH não depende apenas da proximidade física da cromátide irmã, mas também da extensão da ressecção final de DNA17,que inibe o NHEJ.
O reparo de DSB dependente de ecologia inicia-se pela degradação nucleolítica do fio de 5′ das extremidades de ruptura para gerar caudas de DNA de 3′ de fios únicos (ssDNA), um processo chamado de ressecção de 5′-3′. O complexo MRN inicia a ressecção final do DNA e a ressecção adicional é processada em combinação com BLM/EXO1 (proteína da síndrome de Bloom/exonuclease 1) ou BLM/DNA2 (replicação de DNA atp-dependente helicase/nuclease)18,19,20,21,22. A ressecção final do DNA é aprimorada pela CtIP (Proteína Interagindo com ADCBP) através de sua interação direta com o complexo MRN23 e recrutamento do BRCA1 (proteína de suscetibilidade tipo 1 do câncer de mama)24,25. Proteína de replicação A (RPA) se liga prontamente ao ssDNA exposto e, em seguida, é deslocada pela proteína recombinase RAD51 para formar um filamento nucleoproteína que catalisa a busca homólogo e a invasão de fios26,,27,28.
O início da ressecção é um passo crítico para a escolha do caminho de reparo. Uma vez iniciada a ressecção, as extremidades do DNA tornam-se substratos pobres para ligação pelo heterodimer Ku70/Ku80 (componente da via NHEJ) e as células são comprometidas com o HR17,,29,30. O heterodimer Ku70/Ku80 se liga aos fins do DSB, recrutando DNA-PKcs e p53 Binding Protein 1 (53BP1)29,30. O 53BP1 atua como uma barreira à ressecção no G1, bloqueando assim o RH ao promover a NHEJ31,32, mas é removido de forma dependente do BRCA1 na fase S, permitindo, consequentemente, que ocorra a ressecção33,34. Portanto, 53BP1 e BRCA1 desempenham papéis opostos no reparo do DSB, sendo 53BP1 um facilitador NHEJ enquanto o BRCA1 atua permitindo que as quebras sejam reparadas pelo RH.
Em laboratório, a formação de DSB pode ser induzida por radiação ionizante (IR). Enquanto este exemplo utiliza uma alta dose de 4 Gy, 1 Gy e 2 Gy também criam uma quantidade significativa de DSBs, adequado para a análise da formação de focos por proteínas abundantes. É importante notar que o tipo e a dose de radiação utilizada podem levar a diferentes lesões no DNA e na célula: enquanto o IR induz DSBs, também pode causar quebras de fios únicos ou modificação de base (ver35,36 para uma referência sobre transferência de energia linear de irradiação (LET) e tipo de dano de DNA). Para determinar a cinética da formação de focos induzidos por radiação ionizante (IRIF) e sua desobstrução, que indicam reparação dos danos e reversão da formação ativada de DDR8,,9,,37,,38, os focos podem ser monitorados em diferentes pontos de tempo após a radiação ionizante. O tempo de ativação e liberação de todas as principais proteínas de dano de DNA é conhecidocomo 39, e muitos são investigados como marcadores substitutos de eventos-chave. Por exemplo, pRPA, que possui alta afinidade com ssDNA é usado como substituto da ressecção de quebra, proteínas MRN (MRE11, RAD50, NBS1) e exonucleases também podem ser usadas para avaliar a eficiência da ressecção. Enquanto rad51, BRCA1, BRCA2 (proteína de suscetibilidade ao câncer de mama tipo 2) e PALB2 (parceiro e localizador do BRCA2) podem ser monitorados para avaliar a eficiência do RH, a presença das proteínas Ku ou 53BP1, são utilizados como marcadores do NHEJ (Figura 1).
À medida que as proteínas das máquinas de reparação de DNA recrutam umas às outras para a quebra e montagem em superconsetos, as interações DNA-proteína e proteína-proteína podem ser inferidas seguindo sua localização individual ao longo do tempo e analisando a co-localização de proteínas, conforme visualizado por sinais sobrepostos na célula40,,41,,42. Nas linhas celulares, a introdução de mutações pontuais ou exclusão em genes específicos de reparação de DNA, seja através da edição de genomas, ou pela superexpressão de mutantes à base de plasmídeos, permite a investigação de resíduos específicos e seu possível papel no reconhecimento de danos de DNA (por exemplo, co-localização com γH2AX) ou montagem complexa (co-localização com outra, ou várias proteínas), bem como seu impacto na reparação do DNA. Aqui, usamos a imunofluorescência indireta como meio para investigar a formação e resolução de DSBs seguindo os focos γH2AX ao longo do tempo. Também apresentamos um exemplo de formação de focos e análise de co-localização por um dos principais players no reparo de DSB: p53 Binding Protein 1 (53BP1)32. Como mencionado anteriormente, 53BP1 é considerado central para a escolha da via de reparação de DNA. Após o acúmulo de 53BP1 e sua co-localização com γH2AX fornece informações preciosas sobre a fase de ciclo celular, acúmulo de danos de DNA e via usada para reparar DSBs. O objetivo da imunolocalização indireta é avaliar a eficiência da reparação de danos do DNA nas linhas celulares, seguindo o IR como neste estudo, ou após a exposição a várias tensões na célula, desde o cruzamento de DNA até o bloqueio do garfo de replicação (uma lista de agentes nocivos ao DNA é fornecida na Tabela 1).
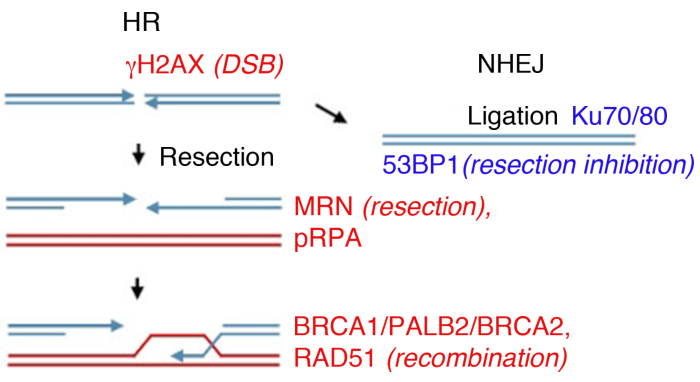
Figura 1: DNA quebra o fio duplo (DSB) caminhos de reparo.
O reparo do DSB envolve duas vias principais: Recombinação Homologous (RH, à esquerda) e End-Joining Não Homólogo (NHEJ, à direita). Após a quebra, as proteínas são ativadas para marcar a quebra (γH2AX), participar da ressecção final (MRN), revestir o ssDNA ressecado (pRPA), promover a recombinação (BRCA1, PALB2, BRCA2, RAD51) ou limitar a ressecção e promover o NHEJ (53BP1). Outras proteínas participam da reparação de danos, mas as proteínas listadas são rotineiramente seguidas por imunofluorescência indireta. Clique aqui para ver uma versão maior desta figura.
| Agente prejudicial ao DNA | Mecanismo de ação | Dose recomendada |
| raios-X | Radiação Formação de quebras duplas com alguns efeitos celulares descontrolados |
1-4 Gy |
| 36 Íons ar | Radiação Formação de quebras duplas encalhadas |
270 keV/μm |
| α-partículas | Radiação Formação de quebras duplas encalhadas |
116 keV/μm |
| Bleomicina | Inibidor da síntese de DNA | 0.4-2 μg/mL |
| Camptothecin | Inibidor de topoisomerase I | 10-200 nM |
| Cisplatina | Agente alquilante (induzir intrastras e crosslinks) |
0,25-2 μM |
| Doxorrubicina | Agente de intercalação Inibidor de topoisomerase II |
10-200 nM |
| Etoposide | Inibidor de topoisomerase II | 10 μM |
| Hidroxiuréia | Inibidor da síntese de DNA (por reductase ribonucleotídeo) |
10-200 μM |
| Metano-metil | Agente alquilante | 0,25-2 mM |
| Mitomicina C | Agente alquilante | 0,25-2 μM |
| Luz ultravioleta (UV) | Formação de dimers de timmidina (gerando distorção da cadeia de DNA) |
50-100 mJ/cm2 |
Tabela 1: Agentes genotóxicos. Exemplos de agentes nocivos do DNA, seu mecanismo de ação e os danos induzidos com base na concentração de trabalho sugerida.
