Using affinity chromatography, recombinant GST-Bla g 1 was readily isolated to a high level of purity (Figure 1A), producing a yield of ~2–4 mg/L of cell culture. Overnight incubation with TEV protease at 4 ˚C is sufficient to remove the GST tag, yielding the final product at ~24 kDa. Note that in this instance there is a significant amount of GST-Bla g 1 in the flow-through and wash fractions, suggesting the Glutathione resin binding capacity was exceeded. The use of more resin or multiple cycles of sample loading and elution could provide remedy for this issue.
Applying the Bla g 1 to a reverse-phase C18 column yields a distinctive elution profile (Figure 1B), with two large peaks at ~50% buffer B, and a second large peak at ~75% buffer B. SDS-PAGE analysis of the resulting fractions suggest that the former correspond to the cleaved GST tag, while the latter corresponds to Bla g 1. Occasionally a third, smaller peak will occur in the middle corresponding to residual, un-cleaved GST-Bla g 1. The presence of this un-cleaved product can be eliminated by increasing the amount of TEV employed in the cleavage reaction or extending the incubation time. While incomplete cleavage will reduce the yield, the separation obtained on the C18 column is sufficient to ensure that the purity of the final Bla g 1 product remains uncompromised. A consequence of reverse-phase HPLC is that the final protein product is eluted into an organic solvent environment. While this facilitates removal of any hydrophobic ligands, removal of this solvent via lyophilization is required, yielding a fluffy white powder (Figure 1C).
Annealing of the protein is required to reconstitute the native Bla g 1 fold and can be carried out either in the absence or presence of a lipid cargo. Addition of DMSO to the dried Bla g 1 and phospholipid cargoes prior to the refolding buffer facilitates the solubilization process, though some longer chain lipid cargoes will not fully dissolve even at elevated temperatures. However, this was not observed to impact the loading efficacy among the lipids tested in our studies (Figure 1C). Similarly, excess lipids will often precipitate out of solution or form large vesicles upon cooling, resulting in a cloudy appearance after annealing (Figure 1C). This was also not observed to effect loading efficiency, and any aggregates are readily removed through the filtration and subsequent buffer exchange steps to yield a clear, transparent solution. Despite the harsh conditions, no thermolysis was observed for Bla g 1.
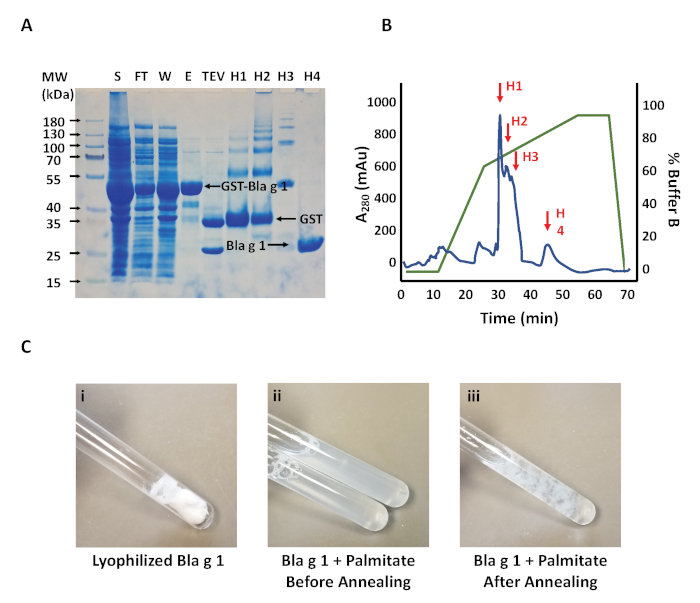
Figure 1: Initial purification of Bla g 1. (A) SDS-PAGE showing the soluble protein fraction following initial lysis (S); flow-through (FT), wash (W), and elution from the glutathione-sepharose column (E); and the final Bla g 1 product following TEV cleavage of the GST tag (TEV). The HPLC elution profile of the resulting Bla g 1 product following TEV cleavage is shown in (B). A280 is shown in blue, while the elution gradient (% Buffer B) is shown in green. Fractions corresponding to the cleaved GST tag (H1, H2), residual un-cleaved GST-Bla g 1 (H3), and purified Bla g 1 (H4) are indicated with red arrows at ~50%, ~65%, and ~74% Buffer B respectively. SDS-PAGE analysis of fractions H1- H4 are shown in (A) and labeled accordingly. (C) Representative images showing Bla g 1 at various stages of the annealing process. Note that the precise and extent of precipitate formation as depicted in ii and iii is dependent on the type of lipid cargo employed. Please click here to view a larger version of this figure.
31P-NMR spectra of Apo-Bla g 1 purified in this manner show no detectable phospholipids either by NMR (Figure 2A) or thin layer chromotography (data not shown). By contrast, similar spectra obtained for Bla g 1 loaded with a distearoylphosphatidylcholine (DSPC) phospholipid show a strong peak corresponding to the phosphatidylcholine headgroup. For comparison, a representative 31P-NMR spectrum of Bla g 1 purified from recombinant E. coli without the use of the lipid removal/annealing protocol described herein (ecBla g 1) show a heterogeneous mixture of endogenous lipids extracted from the recombinant expression system (Figure 2B). Taking advantage of the quantitative nature of NMR, a standard curve can be produced using reference samples of known DSPC concentrations (Figure 2C). Comparing the 31P signal intensity obtained from DSPC-Bla g 1 against this standard curve yields a binding stoichiometry of 4.7 ± 0.5 lipids per protein; a value that compares favorably to the predicted full binding stoichiometry obtained from in silico studies and structural analysis9. Note that this technique will only detect ligands which contain a 31P nucleus such as phospholipids, lysophospholipids, lipopolysaccharides etc. However, this protocol can be easily adapted for 13C-NMR analysis. In this case, methyl-13C labeled fatty acids would be recommended due to its favorable NMR relaxation properties. Restricting isotopic labeling to a single site also facilitates spectral interpretation, as only a single peak is expected, while simultaneously reducing the cost relative to uniform 13C-labeled counterparts. An alternative approach would be to employ mass-spec to identify bound ligands, as demonstrated in a previous study which identified a mixture of fatty acids as the natural cargo of Bla g 1 isolated from cockroach frass (nBla g 1)9. However, the limited quantitation capabilities of mass spec precluded an accurate measurement of binding stoichiometry without sufficient standards.
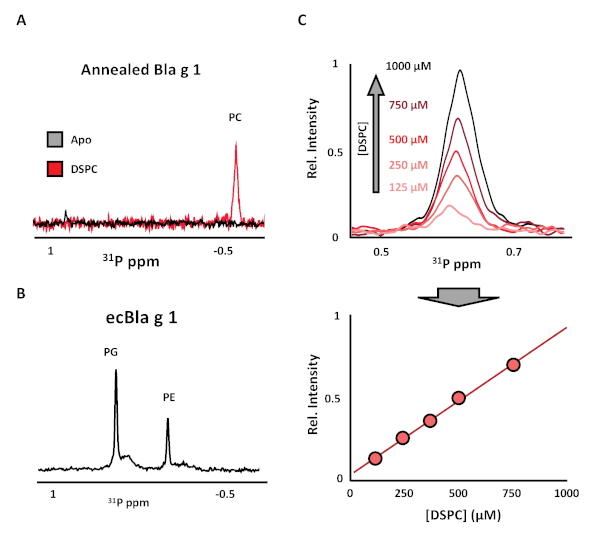
Figure 2: Verifying lipid removal and loading of Bla g 1. (A) 31P-NMR spectra of Apo- (black) or DSPC-loaded Bla g 1 (red) prepared using the annealing protocol described in this work demonstrating the complete removal of lipids in the former, and the homogeneous loading of phosphatidylcholine (PC) lipids achieved in the latter. In contrast, Bla g 1 purified from recombinant E. coli without lipid stripping and annealing (ecBla g 1) shows a heterogeneous mixture of endogenous phosphatidylethanolamine (PE) and phosphatidylglycerol (PG) lipids when analyzed using this method (B). A representative standard curve obtained from DSPC reference samples of known concentrations is shown in (C), from which the Bla g 1 binding stoichiometry can be obtained. Figures adapted from Foo et al. (2019) and presented under the Creative Commons CC BY License9. Please click here to view a larger version of this figure.
Crystal structures of Bla g 1 reveal a unique fold consisting of 12 amphipathic alpha-helices. Circular dichroism represents a quick and convenient method to assess whether this fold has been successfully reconstituted after the annealing process. CD spectra for Apo- and lipid (nMix)-loaded Bla g 1 show minima ~220 and 210 nm indicative of a predominantly alpha-helical structure (Figure 3A). This spectrum is extremely similar to that obtained for ecBla g 1 and nBla g 1, providing further evidence that the native structure of Bla g 1 is successfully recovered. This was further confirmed through the use of 19F and 1H-15N solution-NMR, a full discussion of which is available elsewhere9. CD-based thermal denaturation assays show a cooperative loss of alpha-helical secondary structure indicative of a folded globular domain (Figure 3B). Analysis of the resulting melting temperatures (Figure 3C) show a significant increase upon nMix ligand binding. This elevated thermostability is in line with that calculated for nBla g 1, indicating that we are able to fully reproduce the natural state of Bla g 1. Note that ecBla g 1 also shows a similar, if not greater enhancement in thermostability, illustrating the potential for residual endogenously bound lipids to interfere with biophysical characterization of allergens purified using traditional FPLC-based approaches. In contrast, the ability to quantitatively remove and reload hydrophobic cargoes from allergens such as Bla g 1 provides a unique avenue to examine the role of lipids in the allergic response. Here, we describe a method to examine the influence of lipid cargoes on the structure, stability, and endosomal processing of the allergenic proteins themselves, though other avenues of study could be considered.
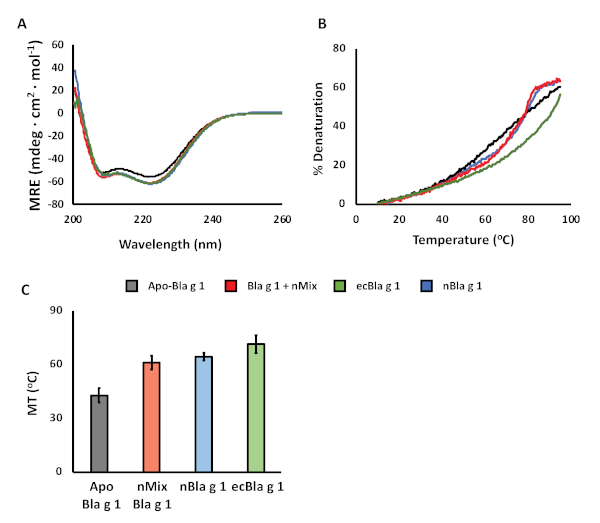
Figure 3: Confirming successful recovery of the Bla g 1 fold. (A) CD spectra of Apo- (black) or nMix-loaded (red) Bla g 1 purified and annealed using the protocol described herein, with minima at ~220 and 210 nm indicative of a predominantly alpha-helical structure consistent with the available X-ray crystal structure. Both Apo- and nMix-loaded Bla g 1 spectra are extremely similar with that obtained for Bla g 1 purified from recombinant E. coli (ecBla g 1, green) or from its natural allergenic source (nBla g 1, blue) without the lipid removal and annealing protocol, further supporting the successful recovery of the native structure in the former. (B) Representative thermal profiles for Apo- (black) and nMix-loaded (red) Bla g 1 showing a sigmoidal curve indicative cooperative unfolding. nBla g 1 (blue) and ecBla g 1 (green) shown as reference. The calculated melting temperatures (MT25) of Bla g 1 are shown in (C). Binding of nMix ligands (red) yields a significant increase in thermostability relative to Apo-Bla g 1 (black). This mirrors the trend observed for nBla g 1 (blue), suggesting that we are able to successfully recover the native state. The even greater stability observed for ecBla g 1 highlights the potential of endogenously bound lipids to interfere with biophysical characterization of allergens. MT25 values presented in C represent the mean value obtained from at least three independent trials. Error bars represent the corresponding standard deviation values. Figures adapted from Foo et al. (2019) and presented under the Creative Commons CC BY License9. Please click here to view a larger version of this figure.
