Over the past decades, advances in stem cell biology and in vitro 3D culture technologies have heralded a revolution in biology and medicine. Higher complexity cell models in 3D have become very popular as they allow cells to grow and interact with a surrounding extracellular framework, closely recapitulating aspects of living tissues including their architecture, cell organization and interactions, or even diffusion characteristics. As such, 3D cell culture models can provide unique insights into the behavior of cells in developing or diseased tissues in vitro. Organoids and spheroids are both multicellular 3D structures, ranging from several micrometers to millimeters, and are the most prominent in vitro 3D structures. Both may be cultured within a supporting scaffold including (i) hydrogels derived from animals (basement membrane extract, collagen), plants (alginate/agarose), or synthesized from chemicals, or (ii) inert matrices containing pores to promote cell proliferation and growth.
Organoids and spheroids can also develop without the presence of a supporting scaffold by relying on cells to self-assemble into clusters. This relies on different techniques such as the use of non-adhesive materials to inhibit cell attachment, surface tension and gravitational force (e.g., hanging drop techniques), or constant circular rotation of vessels (e.g., spinner culture). In all cases, these techniques facilitate cell-cell and cell-matrix interactions to overcome the limitations of traditional monolayer cell culture1. The terms "organoids" and "spheroids" have been used interchangeably in the past, but there are key differences between these two 3D cell culture models. Organoids are in vitro 3D cellular clusters derived from pluripotent stem cells or tissue-specific stem cells, in which cells spontaneously self-organize into progenitors and differentiated cell types and which recapitulate at least some functions of the organ of interest2. Spheroids comprise a broader range of multicellular 3D structures formed under non-adherent conditions and can arise from a large diversity of cell types such as immortalized cell lines or primary cells3. Hence, inherent to their intrinsic stem cell origins, organoids have a higher propensity for self-assembly, viability, and stability than spheroids.
Nevertheless, in essence, these two models are 3D structures composed of multiple cells, and the techniques developed to study them are thus very similar. For example, powerful imaging approaches at the single-cell resolution level are necessary for probing the cellular complexity of both organoids and spheroids. Here, by summarizing this group's expertise and that of leaders in the field of organoids4, this paper describes detailed procedures to perform two-dimensional (2D) and 3D whole-mount staining, imaging, and analyses of the cellular and subcellular composition and spatial organization of organoids and spheroids ranging from 100 µm to several millimeters. Indeed, this procedure presents two different and complementary types of staining and imaging acquisition to analyze a large variety of sizes and types of in vitro 3D cell culture models. The use of one (3D whole-mount analysis) or the other (2D section analysis) will depend on the model studied and the answers sought. 3D whole-mount analysis by confocal microscopy can, for instance, be applied to visualize cells in 3D culture up to 200 µm in depth, irrespective of the overall size of the 3D structure, whereas the analysis of 2D sections provides insights into samples of any size, albeit at the 2D level. This procedure has been successfully applied across a variety of organoids4,5 and spheroids derived from human and murine cells, originating from different embryonic germ layers. The overview of the procedure is shown in Figure 1. The major stages, the relationships between them, decisive steps, and the expected timing are indicated.
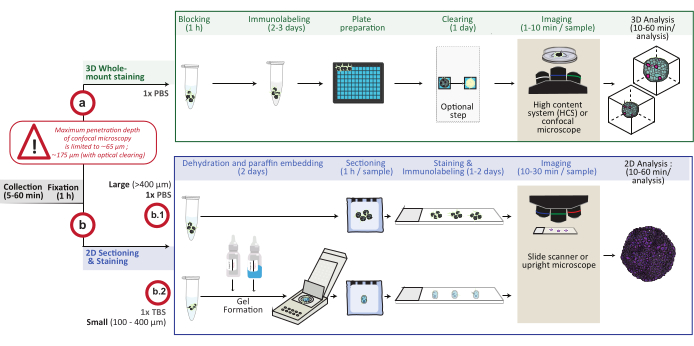
Figure 1: Schematic overview of the procedure. In vitro 3D cell culture models are collected and fixed, then either prepared for 3D whole mount staining (option a) or embedded in paraffin for 2D sectioning and staining (option b). For 3D whole-mount staining experiments, fixed 3D structures are immunolabeled following the fixation step. An optional optical-clearing step can be performed to improve imaging quality and depth of optical microscopy by reducing light scattering during image processing. Images are captured on an inverted confocal microscope or a confocal high content system and analyzed using the appropriate software. For paraffin embedding, 3D structures are directly processed (option b.1 for large structures ≥ 400 µm) or included in a gel (b.2; small structures ≤ 400 µm) for dehydration and paraffin embedding. Paraffin blocks are then cut and stained (histological or immunochemical staining). Images of 2D sections are obtained on a digital slide scanner or an upright microscope and analyzed on an image analysis platform using fast digital quantitative analysis. Please click here to view a larger version of this figure.
NOTE: A loss of ≤25% of the initial number of 3D structures should be expected during the steps involving reagent changes and washing in the following procedure. Plan to use a final number of at least ten 3D structures, with a size ranging from 100 to 500 µm, per tested condition to perform qualitative and quantitative image analyses. If necessary, for larger structures, cut the ends of 1 mL pipette tips to avoid breaking the structures. For all steps, if 3D structure sedimentation is too long, cells can be gently spun at 50 × g for 5 min at room temperature (RT). Depending on the issue investigated, advantages/disadvantages of such a spinning step should be considered, as centrifugation can compromise the shape of the 3D structures. Avoid spinning at >100 × g.
1. Collection and fixation of 3D cell culture models
NOTE: Be careful not to aspirate the 3D structures, which will be only loosely attached to the tube wall.
- Harvesting of 3D cell culture models embedded in a matrix
NOTE: This section describes the recovery of 3D structures grown in drops of a basement membrane extract from murine Engelbreth-Holm-Swarm sarcoma (BME), but may be adapted to other matrices. See the discussion for crucial points regarding ECM.- Remove the culture medium from the wells without disrupting the 3D matrix. Precoat the inside and outside of a 1 mL pipette tip with protein (called precoated 1 mL tip hereafter) by dipping the full length of the tip in 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) (called PBS-BSA 0.1% solution hereafter) and pipetting 1 mL of this solution up and down twice.
NOTE: This precoating will prevent the cells from sticking to the tip and minimize any loss. - Precoat the inside of a centrifuge (15 mL) tube with protein (called precoated centrifuge tube hereafter) by repeatedly filling with PBS-BSA 0.1% solution and emptying the tube.
NOTE: This will prevent the cells from sticking to the tube and minimize any loss. - Using the precoated 1 mL tip, carefully resuspend the 3D structures of the well using 1 mL of ice-cold 1x PBS, and gently transfer the suspension containing the 3D structures to the precoated centrifuge tube.
- Gently add 13 mL of ice-cold 1x PBS, and allow the 3D structures to sediment on ice for at least 10 min.
NOTE: If required, spin for 5 min at 50 × g at 4 °C. Avoid spinning >100 × g, as this will compromise the shape of the 3D structures. - Remove the supernatant. Using a precoated 1 mL tip, gently resuspend the 3D structures in 1 mL of ice-cold 1x PBS. Repeat steps 1.1.4 to 1.1.5 to obtain a homogenous pellet without any 3D matrix residue.
NOTE: Efficient matrix removal is influenced by the type of matrix, the number, and size of 3D structures and requires optimization for different culture conditions. For 3D structures grown in BME, recovery from the matrix removal typically takes 45-60 min. - Using a precoated 1 mL tip, transfer the 1 mL 1x PBS suspension containing the 3D structures to a precoated 1.5 mL centrifuge tube, and proceed with section 1.3.
- Remove the culture medium from the wells without disrupting the 3D matrix. Precoat the inside and outside of a 1 mL pipette tip with protein (called precoated 1 mL tip hereafter) by dipping the full length of the tip in 0.1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS) (called PBS-BSA 0.1% solution hereafter) and pipetting 1 mL of this solution up and down twice.
- Harvesting floating 3D cell culture models
- Using a precoated 1 mL tip, carefully collect and transfer the 3D structures to a precoated 1.5 mL centrifuge tube. Allow the 3D structures to sediment, or spin for 5 min at 50 × g at RT.
- Remove the supernatant. Using a precoated 1 mL tip, resuspend the 3D structures in 1 mL of 1x PBS. Proceed with section 1.3.
- Fixation of 3D cell culture models
- Allow the 3D structures to sediment. Carefully remove the supernatant; under a fume hood, gently resuspend the 3D structures in 1 mL of formalin using a precoated 1 mL tip.
NOTE: Formalin contains formaldehyde, which is hazardous. Manipulate the chemical in a chemical hood. Wear rubber gloves and safety eye goggles. - Incubate the 3D structures for 30 min at RT.
NOTE: A 30 min fixation step with formalin is required for immunostaining of a wide range of 3D structures (varying in size, shape, and origin). However, in general, longer fixation times (>3 h) are better suited to preserve the fluorescence of reporter proteins. - Allow the 3D structures to sediment, or spin for 5 min at 50 × g at RT. Gently remove the formalin, and replace it with 1 mL of 1x PBS. Repeat this washing step in 1x PBS twice. Store the samples at 4 °C, and proceed with section 2 or section 3.
NOTE: The protocol can be paused here, and the cells can be maintained at 4 °C for long-term storage (>1 year).
- Allow the 3D structures to sediment. Carefully remove the supernatant; under a fume hood, gently resuspend the 3D structures in 1 mL of formalin using a precoated 1 mL tip.
2. 3D whole mount staining, imaging, and analysis of 3D cell culture models
NOTE: As the organoids are loosely attached to the tube wall, handle them gently as all following reagent changes can cause sample loss. Before starting, ensure the availability of the correct controls for staining. Positive and negative controls can be cells, in which the protein of interest is known to be either overexpressed or absent, respectively. Incubate samples without the primary antibody to determine if the observed signal is due to non-specific binding of the secondary antibody. As some cells tend to display high levels of autofluorescence, use controls devoid of secondary antibody to determine if the observed fluorescence is coming from background autofluorescence. Immunolabeling and fluorescent reporter visualization can be combined.
- 3D whole mount staining
- Prepare the permeabilization-blocking (PB) solution by supplementing 1x PBS with 0.1%-1% of a non-ionic surfactant (see the Table of Materials), 1% dimethylsulfoxide, 1% BSA, and 1% donkey serum (or from the animal in which the secondary antibodies were raised).
NOTE: Carefully optimize the concentration of the non-ionic surfactant depending on the localization of the target: membrane (0-0.5%), cytoplasm (0.5-1%), and nucleus (1%). This solution can be stored at 4°C for up to 1 month. BSA usually works well for the blocking step, but in case of high background noise, perform an empirical test to obtain the best possible results for a given combination of antibodies. - Transfer the organoids from the 1.5 mL centrifuge tube to a 0.5 mL tube using a precoated 1 mL tip. Let the organoids sediment, gently remove the 1x PBS, and replace it with 0.5 mL of PB solution. Incubate the organoids with gentle horizontal agitation (30-50 rpm) for 1 h at RT.
- Let the organoids sediment, gently remove the PB solution, and wash twice in 1 mL PBS-BSA 0.1% for 3 min.
NOTE: Waiting for 3 min allows the structures to sediment at the bottom of the tube. - Gently remove the PBS-BSA 0.1%, and add 250 µL of primary antibody diluted at the appropriate concentration in PB:1x PBS (1:10) solution. To prepare 10 mL of PB:1x PBS (1:10) solution, dilute 1 mL of PB solution in 9 mL of 1x PBS. Incubate for 2-3 days with gentle horizontal agitation (30-50 rpm) at 4 °C.
NOTE: An appropriate antibody incubation time is crucial for a suitable antibody penetration as 3D structures can sometimes reach large sizes. - Let the organoids sediment, and gently remove the primary antibody solution. Wash 5x in PBS-BSA 0.1% for 3 min per wash and then 2x in 1 mL PBS-BSA 0.1% for 15 min per wash with gentle horizontal agitation.
- Add 250 µL of secondary antibody diluted at 1:250 in PB:1x PBS (1:10) solution. Incubate for 24 h at 4°C with gentle horizontal agitation (30-50 rpm). For this step, protect the samples from light.
- Add 250 µL of Hoechst 33342 (20 µM stock solution) diluted at 1:1000 in PB:1x PBS (1:10) solution, and incubate for another 2 h at 4 °C with gentle horizontal agitation (30-50 rpm).
- Let the organoids sediment, and gently remove the solution containing secondary antibody + Hoechst 33342. Wash the organoids 5x in 1 mL of 1x PBS for 3 min per wash and then 2x in 1 mL of 1x PBS for 15 min per wash with gentle horizontal agitation (30-50 rpm).
NOTE: It is crucial to extensively wash the samples to avoid background noise or loss of signal. - Store the samples in PBS at 4 °C until image acquisition. Proceed with section 2.2.
NOTE: The protocol can be paused here, and the samples can be stored at 4 °C for several months, protected from light.
- Prepare the permeabilization-blocking (PB) solution by supplementing 1x PBS with 0.1%-1% of a non-ionic surfactant (see the Table of Materials), 1% dimethylsulfoxide, 1% BSA, and 1% donkey serum (or from the animal in which the secondary antibodies were raised).
- Sample preparation for confocal imaging
- Using a precoated 1 mL tip, carefully transfer the organoids into 50 µL of the 1x PBS per well in a 96-well black polystyrene microplate. Proceed with step 2.2.3 or section 2.3.
NOTE: At this stage, the sample can be protected from light and stored at 4 °C for many weeks. - Clearing
NOTE: The clearing step is optional and can be used to either immunolabel organoids or to detect endogenous fluorescence. Clearing can cause 3D structure shrinkage, but does not change the general morphology except for spherical mono-layered organoids with large lumens4. For these cystic organoids, skip the clearing step, and perform deep-tissue imaging6.- Prepare 2.5 M glycerol-fructose clearing solution containing 50% v/v glycerol, 11% v/v of distilled water, and 45% w/v fructose by mixing on a magnetic stirrer at least overnight until the solution is completely solubilized and homogenous. Store at 4 °C in the dark for up to 1 month.
- Remove as much 1x PBS as possible without touching the organoids. Add 200 µL of the clearing solution using a 1 mL pipette tip after removing the end, and resuspend gently to prevent the formation of bubbles. Incubate at RT for at least 12 h, and proceed with section 3.
NOTE: As the clearing solution is viscous, small volumes are difficult to handle. To facilitate handling, make sure the solution is at RT, and pipette slowly. For optimal clearing, allow the sample to sediment in the clearing solution for at least 24 h before imaging. If 3D structures are floating at the time of acquisition, perform an optional spin for 10 min at <100 × g at RT, or allow more time (one to several days) to let them sediment. The protocol can be paused at this step before proceeding to imaging if it is protected from light and stored at 4 °C (for weeks) or -20 °C (for months).
- Using a precoated 1 mL tip, carefully transfer the organoids into 50 µL of the 1x PBS per well in a 96-well black polystyrene microplate. Proceed with step 2.2.3 or section 2.3.
- Image acquisition and analysis
NOTE: Image sectioning technology will be required to image 3D structures.- Use confocal microscopes, and favor immersion objectives with higher numerical aperture (NA) compared to air. Choose magnification objectives (10x, 20x, 40x) according to the size of 3D structures, image reconstruction (stitching), and solutions used for the analysis.
- When selecting the acquisition mode, take into consideration the depth of focus of the objective used to define the step for Z stacking; allow for optimal 3D rendering.
NOTE: Image analysis solutions vary, and the analysis will need to be adjusted to the software used. For instance, this analysis protocol was established on a high-content analysis software (see Table of Materials and Supplementary Figure 1 for details) and provides data on object segmentation, calculation of properties, and cell population selection within a 3D reconstructed object.
3. 2D sectioning, staining, imaging, and analysis of 3D cell culture models
NOTE: 3D cell culture models vary in size. Proceed with section 3.1 or 3.2 for efficient paraffin embedding (Figure 2). Allow sufficient time for 3D structure sedimentation before any washes and reagent changes. Be careful not to aspirate the organoids that will be floating at the bottom of the tube. For paraffin embedding, refer to Figure 2 for guidance.
- Paraffin embedding of large (Ø ≥ 400 µm) 3D cell culture models
- On the day before embedding, prewarm two 150 mL flasks filled with paraffin (paraffin baths), a small metal embedding mold per sample, and fine forceps to 65 °C.
- Using a precoated 1 mL tip, carefully transfer the organoids in 1x PBS to a flat-bottom glass tube with a polytetrafluoroethylene-lined bottle cap. Let the organoids sediment, carefully remove the 1x PBS, and replace it with 70% ethanol. Incubate for at least 30 min.
- Let the organoids sediment, and carefully remove the 70% ethanol. Replace it with 1 mL of ready-to-use eosin Y solution. Flick the tube, and stain for at least 30 min. Carefully remove the eosin solution, and dehydrate the organoids in three successive washes with 1 mL of 100% ethanol for ~30 min each.
NOTE: Ethanol, a flammable and volatile liquid, causes severe eye and respiratory tract irritation. Manipulate it in a fume hood, and wear protective eye goggles. - Carefully remove the 100% ethanol, and under a chemical hood, clear the organoids in 3 successive washes with 1 mL of xylene for ~30 min each.
NOTE: Xylene is a toxic, liquid flammable whose vapors may cause irritation. Manipulate it in a fume hood. Avoid direct contact with skin, and wear rubber gloves and protective eye goggles. - Under a chemical hood, prepare a white microtwin tissue cassette by placing a piece of biopsy pad (previously soaked in xylene) inside one of the compartments of the cassette. Carefully transfer the 3D structures using a precoated 2 mL plastic Pasteur pipette to the biopsy pad. Cover them with another biopsy pad soaked in xylene to prevent the organoids from moving, and close the cassette.
- If several samples are processed, place the cassette in a xylene bath to await further processing. Once all samples are transferred into cassettes, place the cassettes in a prewarmed paraffin bath for 30 min at 65 °C. Transfer the cassettes to a fresh prewarmed paraffin bath overnight.
- After paraffin impregnation, take a prewarmed embedding mold, and add the heated paraffin to it. Place the biopsy pad containing the 3D structures into the mold, and gently agitate it until all of the organoids drop to the bottom of the mold. Very carefully place the 3D structures at the center of the mold using prewarmed fine forceps. Proceed with section 3.3.
NOTE: Be careful not to disrupt the 3D structures with the forceps; push, but do not pinch them.
- Paraffin embedding of small (Ø ≤ 400 µm) 3D cell culture models
- On the day before embedding, prewarm two 150 mL flasks filled with paraffin (paraffin baths), a small metal embedding mold per sample, and fine forceps to 65 °C.
- Carefully remove the 1x PBS from the organoid suspension. Gently perform 3 washes in 1 mL of 1x Tris-buffered saline (TBS). Remove as much 1x TBS as possible without touching the organoids.
NOTE: Be careful not to aspirate the sample. If necessary, perform a 5 min spin at 50 x g at RT. Remaining traces of phosphate will interfere with the following steps, notably preventing gel polymerization. Therefore, do not use PBS solutions during any processing step. For this step, a commercial kit, containing cassettes, Reagent #1 (clear fluid), and Reagent #2 (colored fluid), was used to facilitate the paraffin-embedded procedure without potentially loosing tiny fragments (see Table of Materials). Follow kit instructions. The cassettes are preassembled with backing papers and board inserts already in place. - Add 2 drops of Reagent #2 into the tube, and mix gently by tapping the tube. Add 2 drops of Reagent #1, and mix again by tapping to make the gel solidify. Using the fine forceps, remove the gel from the tube, and place it in the well of the cassette.
- Under the fume hood, dehydrate the sample by placing the cassette in successive baths as follows (use the 150 mL flasks, and use fresh ethanol or xylene for each bath): ethanol 70%, 30 min; ethanol 96%, 30 min; ethanol 100%, three washes, 30 min each; xylene, three washes, 30 min each.
- Place the cassettes in a prewarmed paraffin bath for 30 min at 65 °C, and transfer them to a fresh prewarmed paraffin bath overnight. After paraffin impregnation, take a prewarmed embedding mold, and add heated paraffin into it. Open the cassette, carefully dislodge the gel with fine forceps, and place the gel containing the 3D structures onto the center of the embedding mold. Proceed with section 3.3.
- Common steps for paraffin embedding
- Gently transfer the mold to a cold area to let the paraffin solidify in a thin layer, which will maintain the 3D structures in the appropriate position. Add a tissue cassette on top of the mold, and add hot paraffin to cover this plastic cassette. Remove the mold once it is completely solidified, and proceed with section 3.4.
NOTE: Paraffin blocks can be stored at room temperature for years.
- Gently transfer the mold to a cold area to let the paraffin solidify in a thin layer, which will maintain the 3D structures in the appropriate position. Add a tissue cassette on top of the mold, and add hot paraffin to cover this plastic cassette. Remove the mold once it is completely solidified, and proceed with section 3.4.

Figure 2: Overview of the procedure for paraffin embedding of large and small in vitro 3D cell culture models.
(A) Standard procedure for paraffin embedding. After fixation and dehydration, 3D structures are stained with eosin to facilitate their visualization (top and bottom left). 3D structures are carefully placed on the biopsy pad (blue) in the cassette using a 2 mL Pasteur pipet (middle). After paraffin impregnation, the 3D structures are gently dropped into the liquid paraffin using forceps and gently agitated in the biopsy pad. Small 3D structures are lost during this step as they cannot be released from the pad (bottom right: failed embedding). Only large 3D structures will be embedded (top right: successful embedding). Arrowheads point to 3D cultures. (B) Alternative to the standard paraffin embedding protocol. After having fixed small 3D structures, a commercial kit is used to maintain cells in a gel and facilitate their transfer to the mold after paraffin impregnation (right: successful embedding). Please click here to view a larger version of this figure.
- Block sectioning and staining
- Cut 4 µm sections using a standard microtome, and perform standard histological and immunohistochemical techniques. Proceed with section 3.5.
NOTE: Specific slides (see Table of Materials) were used for a better adhesion of sections. The slides can be stored at room temperature or at 4 °C for years.
- Cut 4 µm sections using a standard microtome, and perform standard histological and immunohistochemical techniques. Proceed with section 3.5.
- Image acquisition and analysis
- Perform imaging using a digital slide scanner or upright microscope, and analyze data using a platform for fast digital quantitative analysis that reports morphological and multiplexed expression data on a cell-by-cell basis across entire 3D structure sections (see Supplementary Figure 2 for details).
NOTE: The 20x objective is used routinely by this group.
- Perform imaging using a digital slide scanner or upright microscope, and analyze data using a platform for fast digital quantitative analysis that reports morphological and multiplexed expression data on a cell-by-cell basis across entire 3D structure sections (see Supplementary Figure 2 for details).
This protocol provides an overview of the critical steps for 2D and 3D whole-mount staining, as well as imaging and quantitative analyses of 3D cell culture models (Figure 3 and Figure 4). It is applicable to a wide range of 3D cell culture models-from spheroids to organoids from different host species or tissues-and enables the acquisition of accurate and quantitative information on architecture, cell organization, and interactions at cellular and subcellular levels (Figure 3 and Figure 4). Laboratories may need to optimize 2D histological and immunohistochemical techniques and antibody concentrations according to their own needs.
Both methods yield valuable biological information. 3D whole-mount staining and confocal microscopy provide visual information on cellular composition and spatial position with a field of depth of up to 200 µm (Figure 3B). However, 2D sectioning is convenient for larger 3D structures to reveal detailed cellular morphological traits in the entire section of 3D structures that can be otherwise challenging to observe in situ due to light scattering that compromises resolution in larger samples. Moreover, both techniques can provide quantitative data. Indeed, the resolution obtained allows the application of cellular and subcellular segmentation algorithms for the quantification of the number of cells and the detection of the presence of various cell markers in different cellular subtypes (Figure 3F and Figure 4). In summary, the imaging techniques described here are reproducible, simple, and complementary and represent valuable tools for studying cellular heterogeneity.
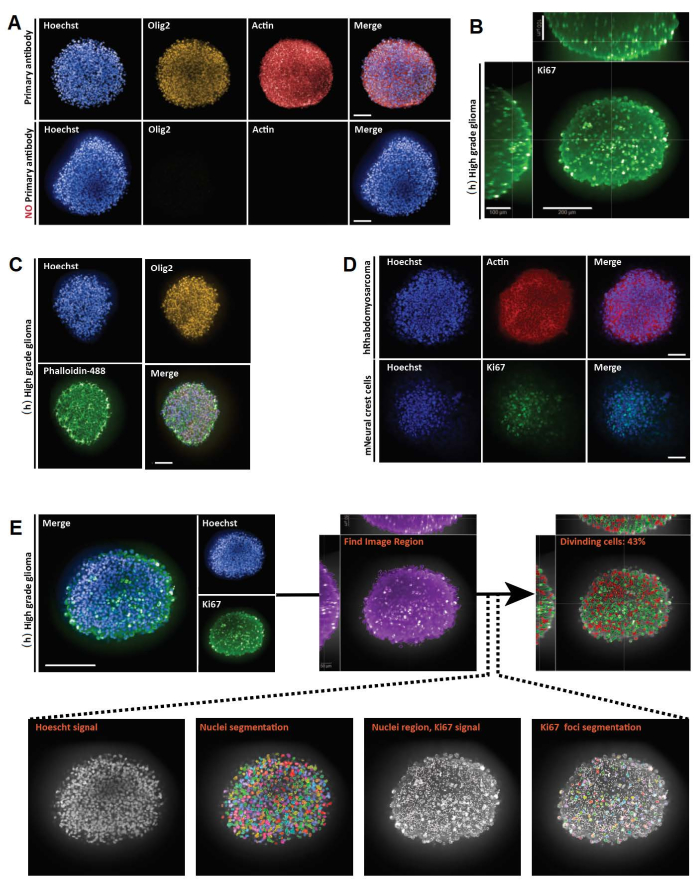
Figure 3: Representative results for 3D whole mount, imaging, and analyses of 3D and 2D optical sections. (A) Confocal images of human (h) high-grade glioma spheroid cultured for a week and labeled with Hoechst (blue), Olig2 (yellow), and Actin (red) (20x water objective). For all acquired images, microscope settings were established using a positive control (top), and then the negative control was imaged using identical settings to control the lack of fluorescence in the absence of primary antibody (bottom). (B) Orthogonal 3D whole-mount representation of Ki67 staining performed in (h) high-grade glioma spheroid cultured for a week (glycerol-fructose clearing; 20x water objective, confocal). (C) Confocal images of (h) high-grade glioma spheroid cultured for a week and labeled with Hoechst (blue), Olig2 (yellow), and Phalloidine-488 (green) (glycerol-fructose clearing; 20x water objective). (D) Confocal images of human (h) rhabdomyosarcoma (top) and mouse (m) neural crest cell (bottom) spheroids cultured for a week and labeled with Hoechst (blue), Actin (red), and Ki67 (green), respectively (glycerol-fructose clearing; 20x dry objective). (E) Confocal images of (h) high-grade glioma spheroid cultured for a week and labeled with Hoechst (blue) and Ki67 (green) (glycerol-fructose clearing; 40x water objective) (top left). Segmented images on the Hoechst channel and Ki67-positive (+) nuclear regions on the green channel were generated using high-content analysis software (see Supplementary Figure 1 and Table of materials) (bottom). Output given is the percentage of Ki67+ nuclei per segmented 3D structure (top right). Scale bar = 100 µm. Please click here to view a larger version of this figure.
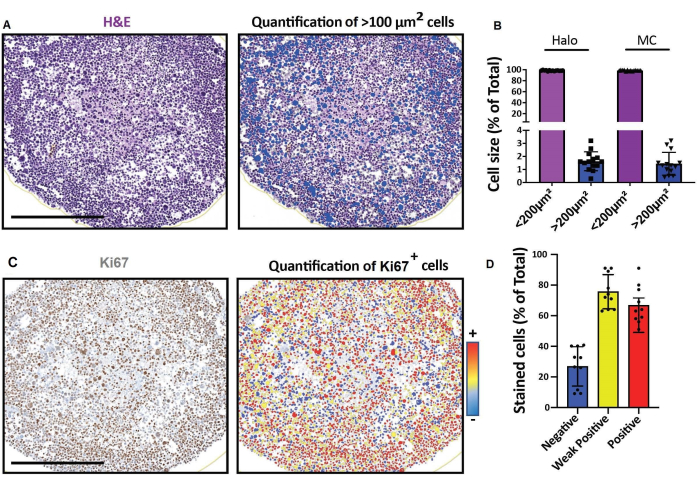
Figure 4: Representative results for imaging and analyses of 2D optical sections. (A, D) 2D section images of a 3D cell model (human rhabdomyosarcoma spheroids cultured for a month) obtained with a digital slide scanner and analyzed on a platform for fast digital quantitative analysis. (A) H&E staining and detection of cells according to their size. Scale bar = 500 µm. (B) Histogram shows percentage of cells < 100 µm2 and > 100 µm2 detected using software for fast digital quantitative analysis (left: Halo) or manual counting (right: MC). (C) Ki67 staining and detection of cells according to the intensity of their 3,3'-diaminobenzidine (DAB) signal. Negative (blue), weakly positive (yellow), positive (red). Scale bar = 500 µm. (D) Histogram shows percentage of Ki67-negative, weakly positive, and positive cells. Abbreviations: H&E = hematoxylin and eosin; MC = manual counting. Please click here to view a larger version of this figure.
Supplementary Figure 1: Overview of the steps in the imaging analysis software. Analyses are based on the association of building blocks. Each building block corresponding to a function-segmentation, calculation, association, output definition-and offers multiple algorithms and variable selections to match the biological sample being imaged. The software provides multiple RMS (Ready Made Solution) analysis protocols that can easily be used and modified. Integrated image analysis protocols can be saved, applied to different datasets, and shared between users. Briefly, the analysis protocol implies sequential object segmentation: spheroid, nuclei and finally, Ki67 pockets (A488). Then, the mean intensity of the Ki67 pockets is calculated to further discriminate the positive events. Finally, nuclei encompassing Ki67 positive pockets are positively selected. Please click here to download this File.
Supplementary Figure 2: Overview of the procedure steps of the quantitative analysis software. Step 1. Upload the files using the Studies tab. Files will be opened in the Image Actions section. Step 2. Open the Annotations tab, then click on Layer Actions to design a new layer all around the structure using the circle tool of the toolbar. For non-circular structures, the pen tool can be used instead. Step 3. The toolbar can be used to design annotations and visualize the quantification with the  tool. Step 4. Open the Analysis tab, and select the best conditions for the analysis of the sample (several trials may be necessary here). Step 4.1. Use the Stain Selection section to set up the staining condition. In the event of several stains, these can be added and renamed, and the virtual color can be modified. The localization detection can be specified-nuclear or cytoplasm staining. Step 4.2. Use the Cell Detection section to set up the cell detection. This section will be the most important for the analysis. The Nuclear Contrast Threshold section will enable detection of all nuclei. Attention must be paid in case there are multiple population sizes, the software can detect several cells instead of a unique big one. Nuclear Size and Nuclear segmentation aggressiveness sections can be used to quantify cell size population ranges. Step 5. Description on how to run sample analysis. Follow steps shown in the figure. Annotation Layer section will run the setting only on this slide. The quantification can be visualized using the tool. Repeat steps 4.1-5 until suitable quantification is achieved. Steps 6-6.1. These steps enable you to draw a figure using the software. Step 7. Quantification graphics obtained via software can be saved. Step 8. Data can be exported. Please click here to download this File.
tool. Step 4. Open the Analysis tab, and select the best conditions for the analysis of the sample (several trials may be necessary here). Step 4.1. Use the Stain Selection section to set up the staining condition. In the event of several stains, these can be added and renamed, and the virtual color can be modified. The localization detection can be specified-nuclear or cytoplasm staining. Step 4.2. Use the Cell Detection section to set up the cell detection. This section will be the most important for the analysis. The Nuclear Contrast Threshold section will enable detection of all nuclei. Attention must be paid in case there are multiple population sizes, the software can detect several cells instead of a unique big one. Nuclear Size and Nuclear segmentation aggressiveness sections can be used to quantify cell size population ranges. Step 5. Description on how to run sample analysis. Follow steps shown in the figure. Annotation Layer section will run the setting only on this slide. The quantification can be visualized using the tool. Repeat steps 4.1-5 until suitable quantification is achieved. Steps 6-6.1. These steps enable you to draw a figure using the software. Step 7. Quantification graphics obtained via software can be saved. Step 8. Data can be exported. Please click here to download this File.
