We used our published deep proteomics datasets25,26,27,30 (Figures 5 and Figure 6) as well as data simulations57 (Table 1) to optimize and evaluate JUMPn performance. For co-expression protein clustering analysis via WGCNA, we recommend utilizing proteins significantly changed across samples as the input (e.g., differentially expressed (DE) proteins detected by statistical analysis). While including non-DE proteins for the analysis may result in more co-expression clusters returned by the program (due to larger input size), we hypothesize that mixing the real signal (e.g., the DE proteins) with the background (the remaining non-DE) for systems-level analysis may dilute the signal and mask the underlying network structure. To test this, simulation analysis was performed under two different conditions: i) highly dynamic proteome (e.g., 50% altered in T cell activation25) and ii) relatively stable proteome (e.g., 2% proteome changed in AD26). For the highly dynamic proteome, six co-expression clusters were simulated from 50% proteome following the same cluster size and expression patterns (i.e., eigengenes) of our published results25. Similarly, for a relatively stable proteome, we simulated three clusters from 2% proteome following our recent AD proteomics study26. As expected, increasing the input number of proteins increases the number of detected clusters (Table 1). For the highly dynamic proteome, using all proteins as input can capture most of the true clusters (5 out of the 6 simulated bona fide clusters; 83% recall) with 63% precision (5 out of the 8 returned clusters are true positives; i.e., the remaining 3 clusters are false positives). However, for the relatively stable proteome, increasing the input size with non-DE proteins dramatically reduces precision (Table 1). For instance, using the whole proteome as input, 169 modules are detected, of which only 2 are correct (1.2% precision; the remaining 98.8% detected modules are false positives). These results thus indicate that choosing only the changed proteome as input will increase the precision of co-expression analysis, especially for relatively stable proteome.
Following the detection of co-expression protein clusters, each cluster will be annotated by JUMPn using the pathway enrichment analysis (Figure 1). The current version includes four commonly used pathway databases, including Gene Ontology (GO), KEGG, Hallmark, and Reactome. Users may also compile their own database in GMT format54, which can be uploaded into JUMPn. Integrating multiple databases for pathway enrichment analysis may provide more comprehensive views; however, the sizes of different pathway databases vary significantly, which may induce unwanted bias to certain (especially large) databases. Two solutions are provided within JUMPn. First, using a statistical approach, nominal p values are adjusted (or penalized) for multiple-hypothesis testing by the Benjamini-Hochberg method58, with a larger database requiring a more significant nominal p-value to reach the same adjusted p level than that from a small database. Second, JUMPn highlights the top significantly enriched pathway for each database separately, thus database-specific top enriched pathways are always displayed.
Similar to pathway enrichment analysis, a composite PPI network was compiled by combining STRING59,60, BioPlex61,62, and InWeb_IM63 databases. The BioPlex database was created using affinity purification followed by mass spectrometry in human cell lines, whereas the STRING and InWeb contain information from various sources. Therefore the STRING and InWeb databases were further filtered by the edge score to ensure high quality, with the cutoff determined by best fitting the scale-free criteria24. The final merged PPI network covers more than 20,000 human genes with ~1,100,000 edges (Table 2). This comprehensive interactome is included and published in a bundle with our JUMPn software for sensitive PPI analysis.
After the analysis is finished, JUMPn generates the publication table spreadsheet file ComprehensiveSummaryTables.xlsx, consisting of three individual sheets. The first sheet contains results of co-expression protein clusterswith one protein per row: the first column indicates the cluster membership of each input protein, and the remaining columns are copied from the user-input file, which contains the protein accession, gene names, protein description, and quantification of individual samples. The second sheet contains results of pathway enrichment analysis, displaying significant pathways enriched in each co-expression cluster. This table is first organized by different pathway databases, then sorted by co-expression clusters, functional pathways, the total number of pathway genes, the total number of genes in the individual cluster, the overlapped gene numbers and names, enrichment fold, Fisher exact test derived P-values and Benjamini-Hochberg false discovery rate. The third sheet contains results of PPI module analysis with one PPI module per row; its columns include the module name (defined by its co-expression membership and module ID, for example, Cluster1_Module1), the mapped proteins and numbers, as well as functional pathways that are defined by searching the module proteins against the pathway databases.
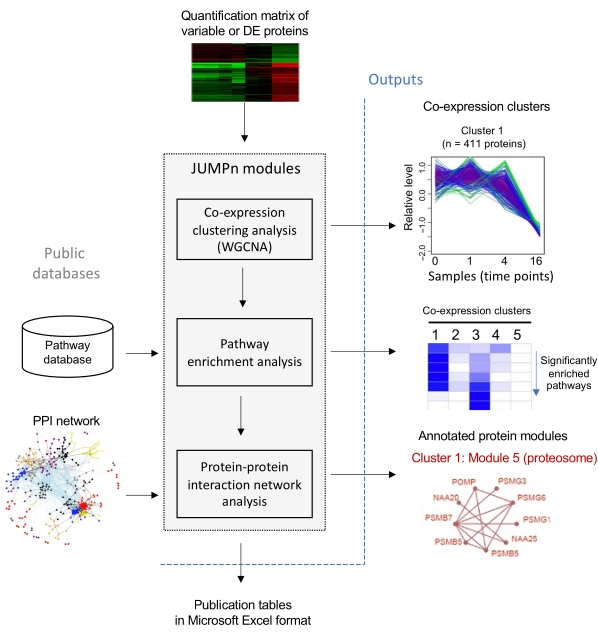
Figure 1: Workflow of JUMPn. Quantification matrix of the top variable of differentially expressed (DE) proteins are taken as input, and proteins are grouped into co-expression clusters by the WGCNA algorithm. Each co-expression is then annotated by pathway enrichment analysis and further superimposed onto the protein-protein interaction (PPI) network for densely connected protein module identifications. Please click here to view a larger version of this figure.

Figure 2: JUMPn welcome page. Please click here to view a larger version of this figure.
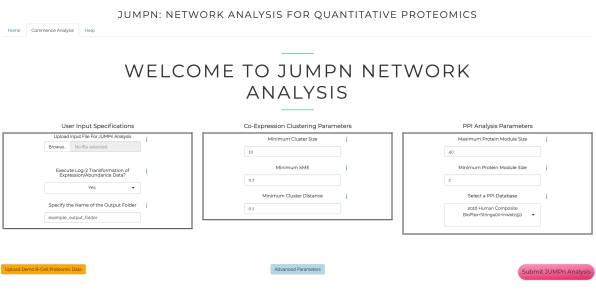
Figure 3: Input page of JUMPn. The page includes the input file upload panel and parameter configuration panels for co-expression clustering and PPI network analysis, respectively. Please click here to view a larger version of this figure.

Figure 4: Example input file of quantification matrix. Columns include protein accession (or any unique IDs), GN (official gene symbols), protein description (or any user-provided information), followed by protein quantification of individual samples. Please click here to view a larger version of this figure.
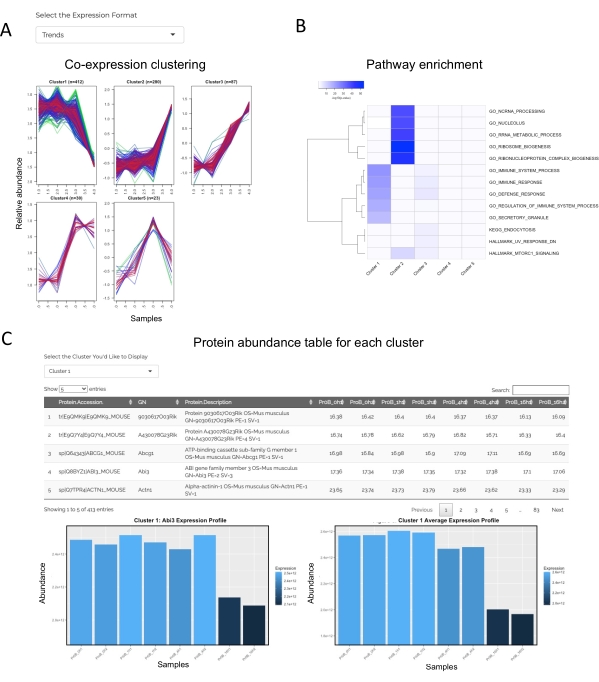
Figure 5: Co-expression cluster results reported by JUMPn. The co-expression clustering patterns (A), top enriched pathway heatmap across clusters (B), and detailed protein abundance for each cluster are shown (C). Users may select various display options and navigate between different clusters via the selection box. Please click here to view a larger version of this figure.
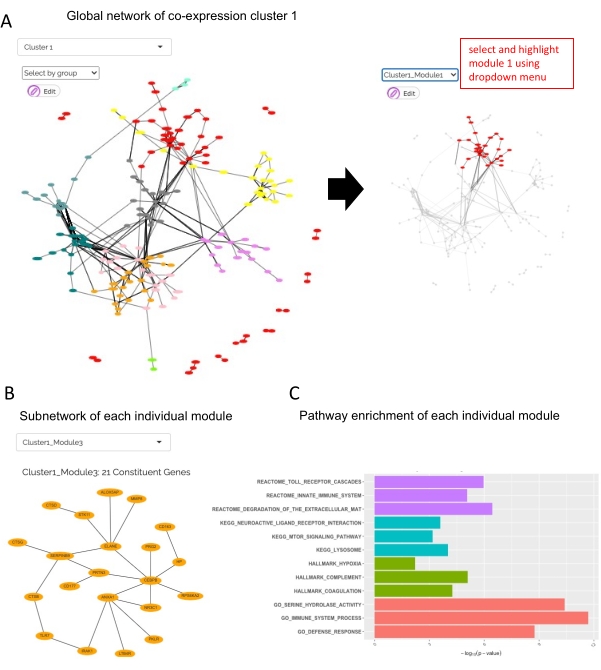
Figure 6: PPI network analysis results reported by JUMPn. The global inter-module network is shown (A), followed by a subnetwork of individual modules (B) and its significantly enriched pathways (C). Users may select various display options and navigate between different clusters and modules via the selection box. Please click here to view a larger version of this figure.
| % top proteins for analysis | # simulated modules | # detected modules | # recaptured modules1 | precision2 | recall3 |
| Highly dynamic proteome (e.g., during T cell activation): 6 simulated modules from 50% proteome | |||||
| 2 | 6 | 2 | 2 | 1 | 0.33 |
| 5 | 6 | 2 | 2 | 1 | 0.33 |
| 10 | 6 | 3 | 3 | 1 | 0.5 |
| 20 | 6 | 4 | 4 | 1 | 0.67 |
| 50 | 6 | 6 | 6 | 1 | 1 |
| 100 | 6 | 8 | 5 | 0.63 | 0.83 |
| Relatively stable proteome (e.g., during pathogenesis of AD): 3 simulated modules from 2% proteome | |||||
| 1 | 3 | 1 | 1 | 1 | 0.33 |
| 2 | 3 | 3 | 3 | 1 | 1 |
| 5 | 3 | 8 | 3 | 0.38 | 1 |
| 10 | 3 | 13 | 3 | 0.23 | 1 |
| 20 | 3 | 19 | 3 | 0.16 | 1 |
| 50 | 3 | 71 | 2 | 0.03 | 0.67 |
| 100 | 3 | 169 | 2 | 0.01 | 0.67 |
| 1A recaptured module is a detected module whose eigengene highly correlates (Pearson R > 0.95) with one of the simulated eigengenes. | |||||
| 2precision = # recaptured modules / # detected modules | |||||
| 3recall = # recaptured modules / # simulated modules | |||||
Table 1: Simulation studies of co-expression cluster detection.
| PPI networks | No. of Nodes | No. of Edges |
| BioPlex 3.0 combined (293T+HCT116) | 14,551 | 1,67,399 |
| InBio_Map_core_2016_09_12 | 17,429 | 6,08,166 |
| STRING (v11.0) | 18,954 | 5,87,482 |
| Composite PPI network | 20,485 | 11,52,607 |
Table 2: Statistics of human protein-protein interaction (PPI) networks. PPI networks are filtered by edge score to ensure high quality, with the score cutoff determined by best fitting the scale-free criteria.
