1. Preparation of methacrylated coverslips
NOTE: Glass coverslips are methacrylated to covalently link PA hydrogels and therefore prevent their detachment during the incubation with cells. Microscope glass coverslips are used to achieve high image quality.
- To prepare a 300 mL solution, take a clean 400 mL glass beaker and place it inside a fume hood.
- Add 10 mL of double distilled water (ddH2O) into the beaker. Add 18.75 mL of acetic acid (≥99.8% pro analysis, p.a.), 18.75 mL of 3-(Trimethoxysilyl)propyl methacrylate and 252.5 mL of ethanol (≥99.8% p.a.) using a serological pipette. Pour the solution into a crystallizing dish.
- Clean 24 mm round glass coverslips with precision wipes. Place the coverslips in a custom-made polytetrafluorethylene rack.
- Immerse the rack into the solution and incubate for 15 min at room temperature (RT).
- Take the rack and rinse all coverslips with ethanol (99.8% p.a.). Dry the coverslips under air flow.
NOTE: Methacrylated coverslips can be stored for up to 1 month at RT.
2. Micropatterning of extracellular matrix proteins on glass coverslips
NOTE: Glass coverslips are first coated with a layer of molecules, comprising of protein and cell repellent. This layer is then removed using a photopatterning technique, to allow the subsequent deposition of extracellular matrix proteins in micropatterned regions.
- Take a 15 mm round glass coverslip and place it on a Petri dish. Use a diamond pen to mark the upper side of the coverslip. Then, proceed to cleaning via oxygen plasma treatment at 0.4 mbar and 200 W for 2 min.
- Pipette 100 µL of 0.01% poly-L-lysine solution on the surface of each coverslip. Incubate for 30 min at RT. Wash the coverslips with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 8.5. Remove any excess liquid but keep the surface wet.
- Pipette 100 µL of 50 mg/mL poly(ethylene glycol) methyl ether succinimidyl valeric acid (mPEG-SVA) in 10 mM HEPES pH 8.5 on the surface of each coverslip. Incubate for 1 h at RT. Rinse the coverslips with 10 mM HEPES pH 8.5 and dry off under air flow.
- Add 2 µL of UV-sensitive photoinitiator (e.g., PLPP-gel) followed by 40 µL of ethanol (≥99.8% p.a.) onto the surface. To obtain a homogeneous distribution of the gel and ethanol on the surface, gently tilt the Petri dish back and forth. Wait for 5 min for the solution to polymerize.
- Place a single coverslip inside a 35 mm Petri dish with a 20 mm hole at the bottom. This allows the laser beam coming from underneath to directly reach the glass surface.
- Turn on the microscope and a light patterning module (working with this device is only permitted after a safety introduction from the laser safety officers). Calibrate the UV-A laser on the 20x air objective. Put the sample with the photoinitiator on the stage. Adjust the focus on the surface of the glass and turn on the focus control. Load and lock the pre-drawn pattern.
- Prepare the pattern using a graphics software like Inkscape. Ensure that the pattern file does not exceed the DMD dimensions (1824 x 1140 px, which corresponds to 552 x 325 µm on 20 x lens (0.28 µm/px)).
NOTE: It is recommended to use the DMD size (1824 x 1140 pixels) as the pattern file size, as this will ensure no error during the patterning. For example, do not produce a pattern file with 1830 x 1130 pixels dimension. - For the purpose of pattern transfer to polyacrylamide, ensure that the patterning is done only up to 60%-70 % of the radius from the center of the glass to the edge.
- For patterning a single cell, use a diameter of 50-100 µm with the distance of 100 µm between patterns and a diameter of 150-300 µm for a cell group. The appropriate pattern size highly depends on the typical cell size, and it has to be sufficiently large to allow cells to adhere.
- Prepare the pattern using a graphics software like Inkscape. Ensure that the pattern file does not exceed the DMD dimensions (1824 x 1140 px, which corresponds to 552 x 325 µm on 20 x lens (0.28 µm/px)).
- Start the patterning by UV dose of 30 mJ/mm2. The patterning duration depends on the number of patterns. Making a single pattern takes approximately 1 s.
- After completing the patterning step, rinse the surface with phosphate-buffered saline (PBS) three times. Incubate the sample with 100 µL of 25 µg/mL fibronectin dissolved in 1x PBS and 25 µg/mL fibrinogen Alexa488 conjugate in PBS for 1 h at RT. For other ECM proteins, find the optimal concentrations completely covering the patterned regions.
- Rinse the surface with 1x PBS three times. Ensure that the patterned glass is immediately used for glass-PA transfer. Store the patterned glass in PBS at RT during the hydrogel preparation.
3. Fabrication of patterned polyacrylamide hydrogels
NOTE: Polyacrylamide hydrogels are prepared, including oxidized N-hydroxyethyl acrylamide (HEA) to present reactive aldehyde groups for the covalent binding of matrix proteins on the surface. Additionally, a dual-layer approach to embed fluorescent beads only on the top layer of the hydrogel is used to improve the recording of bead displacement during traction force microscopy experiments.
- Put the methacrylated glass coverslips into a Petri dish.
- To prepare fresh oxidized HEA solution, add 9.55 mL of double-distilled water into a 15 mL centrifuge tube. Then, add 0.5 mL of HEA, and 42 mg of sodium(meta)periodate to get 10 mL of the solution. Continuously stir the solution in the dark for 4 h.
- Mix acrylamide, bis-acrylamide, and double-distilled water according to Table 1, to get 10 mL of stock solution hydrogel (do it under the fume hood, monomers are neurotoxic). Degas and close the lid to an airtight seal.
NOTE: The stock solution can be kept in aliquots for up to 1 year at 4 °C. Always characterize the Young's modulus of the hydrogel prior to use. - Prepare a dual-layered PA hydrogel (do it under the fume hood, monomers are neurotoxic).
- Start with the bottom layer by gently mixing 99.3 µL of stock solution, 0.5 µL of 1% ammonium persulfate (APS), and 0.2 µL of tetramethylethylenediamine (TEMED) in a 1.5 mL tube. Take 10 µL from the solution and pipette it dropwise on the center of the methacrylated coverslip.
- Carefully place a 15 mm round coverslip on the droplet and wait for 45 min for polymerization. Gently detach the top coverslip with a scalpel.
- For the top layer, gently mix 93.3 µLof stock solution with 1 µL of fresh oxidized HEA solution, 5 µL of fluorescent beads (corresponding to three beads per square micron for beads of 200 nm sized beads), 0.5 µL of 1% APS, and 0.2 µL of TEMED in a 1.5 mL tube. Take 5 µL from the solution and pipette it dropwise on the center of the already polymerized bottom layer.
- Gently place the micropatterned coverslip on the droplet. Wait for 45 min at RT for the droplet to polymerize. Make sure the patterned side touches the droplet. Gently detach the coverslip with a scalpel.
- Glue the 24 mm coverslips to the bottom of custom-drilled 6-well plates using two-component silicone-glue. After 5 min, add PBS to the wells.
- Characterize the Young's modulus of the polyacrylamide hydrogel samples by atomic force microscopy (AFM).
- Mount a spherical tip cantilever on the AFM holder.
- Calibrate each cantilever by acquiring a force-distance curve (3-4 V setpoint) on a hard substrate (e.g., glass or mica), extrapolate the cantilever sensitivity, retract from the surface, and perform a thermal tune to get their spring constant.
- Place the calibrated cantilevers over the hydrogel sample and acquire force curves in PBS buffer, using the following parameters: 20-30 nN force setpoint, 5 or 10 µm/s approach and retraction speed, 5 µm ramp size.
NOTE: An inverted optical microscope, equipped with an air 40x objective and a green fluorescent protein (GFP) filter was used to visualize and target ECM-micropatterned areas within the PA hydrogel samples. - Plot the acquired force versus distance curves with the AFM analysis software.
- Find the contact point and convert force versus distance curves into force versus indentation curves.
- Fit the indentation part of the curve with Hertz model (or a suitable mechanics model in function of the tip geometry), using a Poisson ratio between 0.2 and 0.5.
4. Local release of cell traction forces by UV-A laser illumination (LUVI-TFM)
NOTE: UV-A laser is applied to release traction forces in cells localized in defined regions of the hydrogels. The UV-A laser (i.e., λ = 375 nm solid-state laser, <15 mW) is a class 3B laser. The unshielded laser beam is dangerous to the eyes and often for the skin. Reflected and scattered light and radiation can be dangerous. Moreover, UV radiation may cause skin cancer. Working with devices is only permitted after safety introduction from the laser safety officers.
- Aspirate the PBS from the wells.
- Seed 3 x 106 cells per 6-well plate in growth medium. For fibroblasts, use high glucose Dulbecco's Modified Eagle Medium (DMEM) containing L-glutamine and supplemented with 10% Foetal Bovine Serum (FBS) and 1% penicillin/streptomycin. Incubate the cells overnight at 37 °C/5% CO2. Wash samples to remove floating cells before starting microscopy.
- Turn on the microscope incubation chamber and gas supply to obtain a temperature of 37 °C and 5% CO2 atmosphere.
- Turn on the microscope and the laser patterning module. If necessary, re-calibrate the UV-A laser on the 20x air objective using the Leonardo software. Place the custom-made 6-well plate with the samples on the stage. Adjust focus on the surface of the PA.
- Setup the illumination pattern and mark the cells of interest. Refocus on the surface of the hydrogel. Acquire the image of the cells in the brightfield channel. Switch to the Cy5 channel (far-red filter, 650 nm excitation, 670 nm emission) and take an image of beads in the deformed state.
- Shift to the laser channel. Turn on the laser for 3 min. In our particular setup, this corresponded to a light dose of 6,000 mJ/mm2.
- Switch to the Cy5 channel and acquire an image of the beads in the undeformed state.
NOTE: For experiment using mouse embryonic fibroblasts, wait for 15 min post-exposure to record the reference/undeformed image (See the Representative Results section). It might be different for other type of cells. Repeat steps 4.5-4.7 again for another cell(s) of interest. - To measure the elevated oxidative stress after UV-A illumination, use the commercially available reagent (CellRox). CellRox is non-fluorescent in the reduced state and, when oxidized by reactive oxygen species, fluoresces with an emission maximum of ~665 nm.
- According to the provided protocol, add 5 µM reagent into the imaging media and incubate for 30 min at 37 °C/5% CO2. Then, wash the cells 3x with warm 1x PBS and replace the imaging media. Record the Cy5 signal by fluorescence imaging before and after UV-A illumination using a similar light exposure.
5. Image processing, particle image velocimetry and calculation of traction forces
NOTE: Cell traction forces are recorded by analyzing displacement of fluorescent beads and calculated using image analysis tools based on particle image velocimetry.
- Open fluorescent beads images, before (i.e., deformed) and after laser (i.e., undeformed) using Fiji. Merge two images as one stack (Image | Stacks | Images to Stack).
- Correct lateral drift between the two images using StackReg plugin (P. Thévenaz, Swiss Federal Institute of Technology Lausanne). Change images to 8 bit (Image | Type | 8 bit) and re-scale to 1024 x 1024 pix (Image | Scale). Save as an Image Sequence (TIFF format) with the reference image always at the beginning.
- Acquire the displacement vector field using a cross-correlation technique19 from the PIV-field as implemented in the OpenPIV project.
- Ensure that the relaxed (undeformed) image and the deformed image are covered with search windows of size wR and wD in pixels. For each search window, define a cross-correlation function using the following formula:
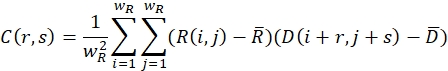
Here, R(i,j) and D(i,j) describe the intensity fields of the two images truncated to the selected search window and subsequently mirror padded.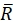 and
and 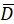 describe their mean value. The window sizes are chosen to be wR = 32 and wD = 32 and an overlap of 70% is used between the neighboring search windows.
describe their mean value. The window sizes are chosen to be wR = 32 and wD = 32 and an overlap of 70% is used between the neighboring search windows. - For each search window determine a displacement vector
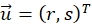 that optimizes the cross-correlation function and assign to the center position of the search window. Subpixel accuracy is obtained using a Gaussian fit around the maxima region as described20.
that optimizes the cross-correlation function and assign to the center position of the search window. Subpixel accuracy is obtained using a Gaussian fit around the maxima region as described20. - Find and remove ambiguous displacement vectors. Displacement vectors corresponding to search windows where the ratio between two highest local maxima of the cross-correlation function below a threshold of 1.5 are considered as ambiguous21.
- Discard displacement vectors that fail the normalized median test for a residual threshold of 1.522.
- (Optional) Correct the remaining lateral drift by subtracting a fixed vector
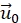 from all displacements. This is done because the displacement in a selected area far away from the cell vanishes.
from all displacements. This is done because the displacement in a selected area far away from the cell vanishes. - Interpolate the resulting displacement vector field to a regular grid with spacing of 4 px of the input images using a cubic polynomial interpolation. Missing datapoints are filled using a smooth bivariate spline extrapolation. A displacement vector in pixels is related to a local deformation by the pixel ratio of the image (0.33 µm/px for 20x air lens).
- (Optional) Mirror pad the image to reduce ringing artifacts in the subsequent analysis.
- Multiply the deformation field by a Tukey window function to eliminate the edge effect due to the drift correction, with a parameter of 0.2-0.3. In this experiment, the micropatterned area which is in the center of FOV is of interest.
- Ensure that the relaxed (undeformed) image and the deformed image are covered with search windows of size wR and wD in pixels. For each search window, define a cross-correlation function using the following formula:
- Reconstruct the traction using regularized Fourier Transform Traction Cytometry (FTTC)18. As regularization we use the simplest reasonable choice, namely, 0th order Tikhonov (L2) regularization23,24,25. An optimal regularization parameter λ is chosen such that it minimizes the Generalized Cross Validation (GCV) function defined by26:
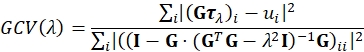
Here τλ is a stacked vector containing the x and y components of the reconstructed traction field for the given value of λ for all Fourier sampling modes and G is the linear operator mapping traction fields to their corresponding displacement field, as defined for FTTC. The sum is running over vector components. ui are the x and y components of the displacement field for all Fourier sampling modes. GCV is widely used in the field of ill-posed inverse problems and is a good alternative to the L-curve criterion often used in TFM. A Lanzcos filter is used to reduce artifacts introduces due to the limited frequency space and the displacement field upsampling. The traction calculation depends on the Young's modulus of the PA (e.g., 11.3 kPa) and Poisson's ratio (e.g., for PA in the range between 0.2 and 0.5 depending on crosslinker concentration).
The PA hydrogels were polymerized on methacrylated glass coverslips, which present reactive groups for the covalent linkage of the PA. In this way, when placing the hydrogels in an aqueous solution, their detachment from the substrate was prevented (Figure 1A–D). To obtain a high density of fiducial markers near the hydrogel surface, we developed the preparation of a dual-layered PA hydrogel. The bottom layer, without any fluorescent beads, was polymerized on the methacrylated coverslip. Subsequently, another layer containing beads was polymerized on top of the bottom layer (Figure 1E–H), replacing the need for sedimentation, which is often used to achieve bead distribution in a single focal plane and increase bead density. To achieve control over cell shape during TFM measurements, we produced micropatterned PA hydrogels via direct transfer of fibronectin-patterned microstructures from a glass coverslip to the pre-polymerized PA (Figure 1F–F'). The addition of 1% non-conventional crosslinker (oxidized HEA) in the top hydrogel layer solution provides aldehyde groups that covalently bind to amine groups of fibronectin.
We prepared PA hydrogels, which are approximately 60 µm in thickness and confined fluorescent beads in the upper layer close to the hydrogel surface. This type of hydrogel is a suitable substrate for imaging cellular tractions with inverted microscopes and thick enough (minimum thickness to perform TFM has been reported to be 20-30 µm)18 to prevent any impact of the glass substrate. Localization of the fluorescent beads was imaged by confocal microscopy (Figure 1I). To visualize protein transfer on the hydrogel, we used fluorescently labeled ECM proteins and imaged them by epifluorescence microscopy (Figure 1J). We prepared micropatterned circles of fibronectin with a diameter of 100 µm (left side) and 50 µm (right side) to allow adhesion of groups of cells or single cells, as shown in the overlay of bright field images of adherent cells, and fluorescently labeled fiducial beads and fibronectin micropatterns. On a different sample, the adhesion response of single cells to micropatterned fibronectin was validated by indirect immunofluorescence microscopy to image the localization of focal adhesion proteins such as paxillin (Figure 1K).
Both ECM protein coating and hydrogel stiffness are crucial parameters for cell adhesion26. To characterize the mechanical properties of our PA hydrogels, we performed nanoindentation experiments by AFM27, coupled with an epifluorescence microscope. Hydrogel substrates of three different stiffnesses were prepared and tested with our setup (Figure 2 and Table 1). Sphere-shaped AFM cantilevers were cyclically approached and retracted from unpatterned PA hydrogels and fibrinogen conjugated to Alexa488-micropatterned areas, while recording force-distance curves. Subsequently, contact point evaluation and application of Hertz model allowed us to estimate the Young's modulus (E) of each sample. ECM micropatterning did not alter the Young's modulus, which remained comparable to each of the unpatterned PA hydrogels.
To measure traction forces exerted by adherent cells on the substrate, we developed an experimental setup for reference based TFM carried out on a widefield epifluorescence microscope equipped with near UV laser module (UV-A 6000 mJ/mm2) to release cell tractions (Figure 3A). As the laser beam illumination can be spatio-temporally controlled, not only is it possible to selectively expose a single cell or a cell cluster with a high laser dose, but also and more importantly, the disruptive intermediate step of cell removal from the entire sample using a digestive enzyme like trypsin is no longer necessary.Illuminating cells with such a high laser dose induced an elevated oxidative stress leading to cell death (Figure 3B). Cell death yielded the release of tractions from the substrate, as indicated by the bead displacement (illustrated in Figure 3A). We combined the UV-A illumination with a light patterning module to selectively illuminate micron-sized regions of the PA hydrogels (Figure 3B–C). In this way it is possible to release the tractions of micropatterned cell clusters (Figure 3C,F) or single cells (Figure 3B). Importantly, at our time scales the mechanical properties of the ECM patterned hydrogels were not significantly affected by UV exposure (Figure 2C). Increase in oxidative stress is shown in Figure 3D,E. The traction forces were reconstructed by using regularized FTTC with a regularization parameter chosen by Generalized Cross Validation (Figure 3B,F). The release of forces for single cells occurred over a period of 15 min, and the result of the LUVI-TFM is comparable to the trypsin-based TFM (Figure 3G–H).
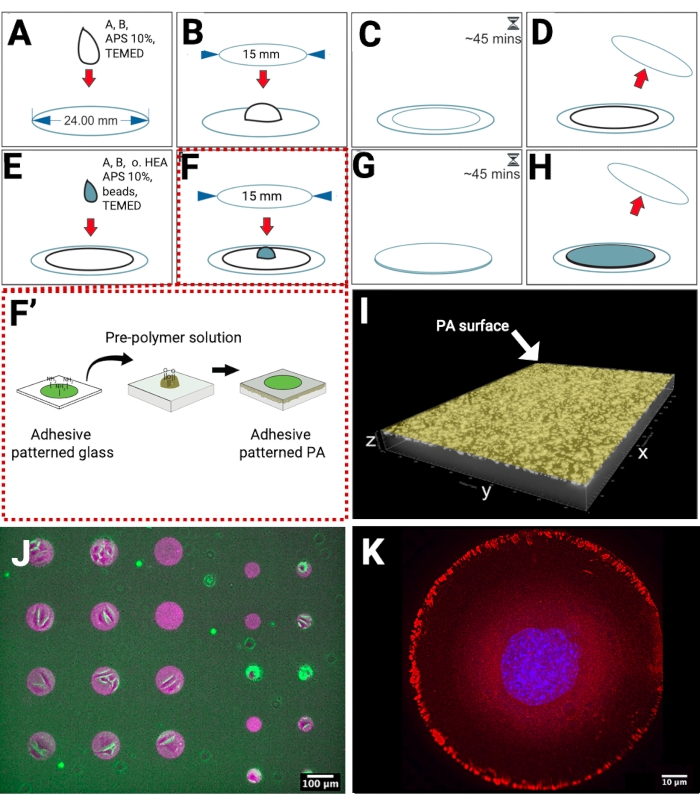
Figure 1: Scheme of patterned fibronectin-substrate preparation for TFM. (A) The solution for the bottom layer is pipetted on a methacrylated surface. (B) A smaller clean coverslip is carefully placed on the droplet. (C) The gelation time is 45 min. (D) The top coverslip is detached. The bottom layer is ready. (E) The solution for the top layer is pipetted on the hydrogel. (F) The patterned coverslip is carefully placed on the droplet. (F') The micropatterning of adhesive proteins on glass is produced by maskless near UV-lithography, and then transferred from glass to PA hydrogel. The free amine groups of adhesive proteins, e.g., fibronectin, bind covalently to aldehyde groups on the PA surface. (G) The gelation time is 45 min. (H) The top coverslip is detached. The patterned PA hydrogel is ready. (I) A 3D representation of a confocal xyz micrograph showing a high density of fiducial beads near the surface. (J) A micrograph of fluorescently labeled fibronectin (magenta) on the surface of PA with embedded fluorescent beads (green), overlayed with a bright field image of cells. (K) Indirect immunofluorescence microscopy imaging of a single cell adhering to a circle-shaped fibronectin micropattern (100 µm). Nucleus (blue), paxillin (red). Please click here to view a larger version of this figure.
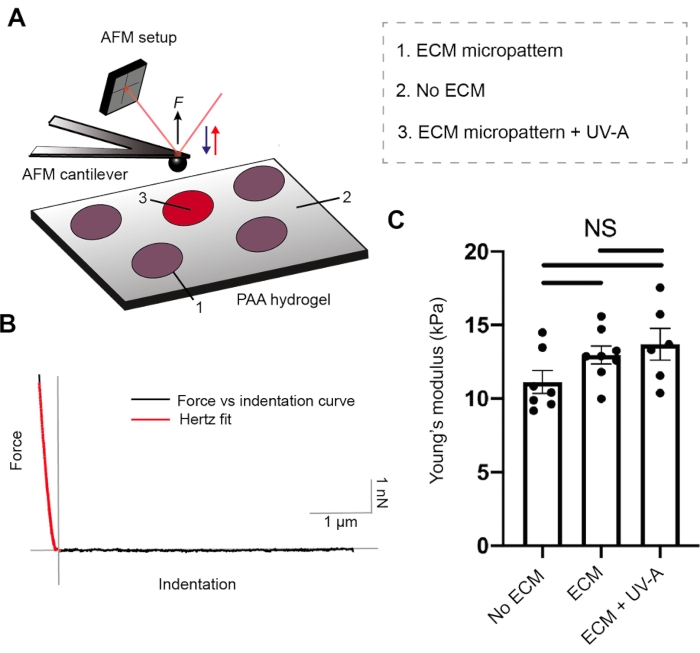
Figure 2: Characterization of sample mechanical properties by AFM. (A) AFM nanoindentation experimental setup. A sphere-shaped cantilever is used to probe ECM micropatterns before or after UV treatment, and the non-patterned regions of dual layered PA (5% Acrylamide, 0.3% Bis-acrylamide) areas. (B) Representative force versus indentation curve for ECM micropattern (black). Hertz fit (red) is used to calculate the Young's modulus (E) of the sample. (C) Mechanical measurements for non-patterned dual layered PA areas (no ECM), ECM micropattern and ECM micropattern after UV-A exposure. The bars show mean ± S.E.M. (Brown-Forsythe and Welch ANOVA test). Please click here to view a larger version of this figure.
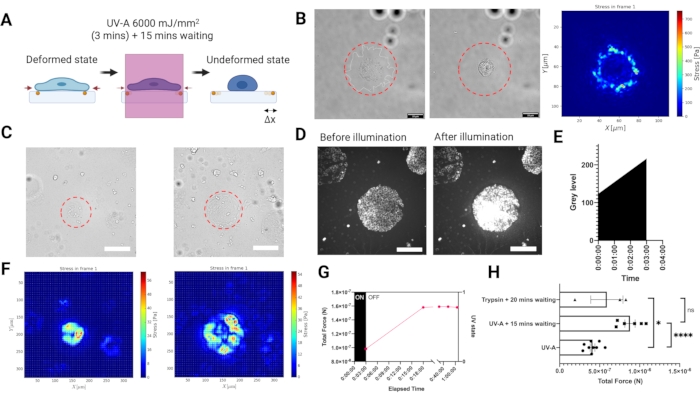
Figure 3. Local UV-A illumination TFM (LUVI-TFM) enables local traction force measurements inside a large field of view. (A) Scheme of local UV-A illumination TFM. Cells are treated with a high dose of UV-A laser to get the undeformed (reference) image. (B) The brightfield image of a single cell before UV-A laser illumination. Middle: The brightfield image of a single cell after UV-A laser illumination. Right: Traction force of a single MEF adhering to a fibronectin coated PA hydrogel (E = 5.74 kPa) illuminated with a 50 µm diameter UV-light beam (red dashed line). (C) Left: Recording of traction forces of a cluster of mouse embryonic fibroblasts (MEF) adhering to micropatterned fibronectin (circle, 100 µm diameter). The cluster was illuminated with a 200 µm diameter UV-light beam (red dashed line). Right: Recording of traction forces of a cluster of mouse embryonic fibroblasts (MEF) adhering to micropatterned fibronectin (circle, 300 µm diameter). The cluster was illuminated with a 300 µm diameter UV-light beam (red dashed line). Scale bar = 200 µm. (D,E) An increase in oxidative stress in cells indicated by the increased Cy5 signal intensity after a high UV-A laser dose is detected by fluorescence microscopy. The oxidative stress leads to cell death. Scale bar = 200 µm. (F) Left: The stress heat map from a cluster of mouse embryonic fibroblasts (MEF) adhering to micropatterned fibronectin (circle, 100 µm diameter). Right: The stress heat map from a cluster of mouse embryonic fibroblasts (MEF) adhering to micropatterned fibronectin (circle, 300 µm diameter). (G) MEF cells adhering to 100 µm fibronectin micropatterns slowly release their tractions after UV-A exposure. Full release is achieved after 15 min (the reference image was then taken 15 min post exposure). (H) A comparison with the conventional trypsin based-TFM for MEF cells adhering to 300 µm diameter patterned-fibronectin. The result of UV-A illumination (6,000 mJ/mm2) followed by 15 min waiting is non-significantly different from the conventional trypsin treatment (i.e., 0.05% for 20 min). The bars show mean S.E.M. (two-tailed Student's t-test, ****P < 0.0001, * P < 0.05, ns P ≤0.5). Please click here to view a larger version of this figure.
| Acrylamide (%) | Bis-acrylamide (%) | Acrylamide from 40 % stock solution (ml) | Bis-acrylamide from 2 % stock solution (ml) | Water (ml) | Young’s modulus (kPa) |
| 4 | 0.1 | 1 | 0.5 | 8.5 | 5,74 ± 0,53 |
| 5 | 0.15 | 1.25 | 0.75 | 8 | 9,69 ± 0,68 |
| 5 | 0.3 | 1.25 | 1.5 | 7.25 | 11,33 ± 1,06 |
Table 1: Hydrogel stock solution mixtures and resulting elasticity
All data are deposited in the Open Access Data Repository of The Max Planck Society (Edmond) and can be accessed through the following address: https://edmond.mpdl.mpg.de/imeji/collection/JTu8PlWqpbymN9Qf
