Isolation of cells and tissue from animals was in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and approved by the Wake Forest Institutional Animal Care and Use Committee
NOTE: This protocol is used on samples already treated and fixed per specific experimental paradigms and requirements. For demonstration purposes, synapse formation due to rapid antidepressant treatment is used to highlight this synapse detection technique6. Neurons previously cultured on coverslips, treated, fixed in 4% paraformaldehyde (PFA), and stored in 1x phosphate-buffered saline (PBS) will be used to highlight the in vitro procedures. Previously sliced hippocampal tissue (25 µm thick) from mice treated, transcardially perfused with ice-cold PBS and 4% PFA, and then stored in cryoprotectant will be used to highlight the slice procedures. Please see11,12 for more information about how to culture neurons or transcardially perfuse rodents. See Figure 1 for a graphical representation of this procedure.
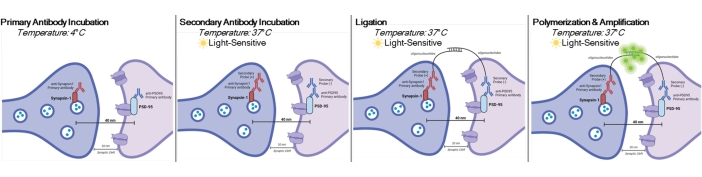
Figure 1: Graphical representation of DetectSyn assay. After permeabilizing cell membranes, primary antibodies for Synapsin1 and PSD95 bind to these synaptic proteins. Secondaries with oligonucleotide tags then bind to the primary antibodies. If Synapsin1 and PSD95 are within 40 nm, as at a synapse, then the oligonucleotides interact, and a fluorescent tag is amplified. This fluorescent signal can then be imaged via standard microscopy and analyzed. Please click here to view a larger version of this figure.
1. Rinse samples
- Rinse the samples with 500 µL of 1x PBS + 0.75% glycine for 5 min 3 times with gentle agitation on an orbital shaker to remove residual PFA or cryoprotectant.
2. Block and permeabilize samples
- Prepare blocking and permeabilization solution (10% normal donkey serum, 0.25% Tween 20) in 1x PBS. Prepare enough to use for blocking, primary, and secondary incubations.
- To samples (e.g., coverslips or free-floating slices) in 24 well plates, add 500 µL of blocking and permeabilization solution. Ensure each well contains a different sample and is appropriately labeled to prevent samples from being switched.
- Incubate the samples at room temperature (RT) for 60 min for cultured cells or 2 h for sliced tissue. Use an orbital shaker for gentle agitation.
3. Incubate samples in primary antibodies
- Prepare primary antibodies in blocking buffer:
- Prepare Postsynaptic density 95 (PSD95; 1:500, rabbit polyclonal), Synapsin1 (1:500, mouse monoclonal), MAP2 (1:400, chicken polyclonal)
- Prepare a negative control aliquot that omits one of the synaptic pairs (e.g., without PSD95)
- Carefully remove the blocking solution with a plastic Pasteur pipette. Try to remove as much as possible without disturbing the cells or tearing tissue.
- For cultured cells:
- Line a large plastic petri dish with parafilm. Carefully transfer coverslips to the parafilm using forceps.
- Carefully add 60 µL of the primary antibody solution to the top of the coverslips. Make sure not to spill the primary antibody solution over the side of the coverslip.
- To provide humidity and prevent samples from drying out during the incubation period, add ultrapure water to a smaller Petri dish and carefully arrange the small Petri dish around the coverslips.
- Cover the large Petri dish and incubate cultured cells for 1 h at RT.
- For sliced tissue:
- Carefully add 250 µL of the primary antibody solution to free-floating slices in a 24 well plate.
- Cover the plate and incubate the tissue overnight at 4 °C with gentle agitation on an orbital shaker.
4. Wash samples, then incubate in secondary antibodies
- Prepare secondary antibodies in the blocking buffer:
- Prepare Donkey anti-mouse (1:5), donkey anti-rabbit (1:5), donkey anti-chicken (1:400).
- At this step, additional technical controls can be obtained by preparing a secondary aliquot that omits either the anti-mouse or anti-rabbit secondary.
- For cultured cells:
- Using forceps, carefully tap off the primary solution from coverslips onto a paper towel
- Using forceps, carefully transfer the coverslips back to their original 24 well plate filled with 500 µL of 1x PBS.
- For sliced tissue:
- Carefully remove the primary antibody solution with a plastic Pasteur pipette. Try to remove as much as possible without tearing tissue.
- Add 500 µL of 1x PBS
- Wash the samples for 10 min 3 times in 1x PBS with gentle agitation on an orbital shaker. During this time, bring all wash buffers to RT.
- During this time, change the parafilm in the large Petri dish
- For cultured cells:
- Using forceps, carefully transfer the coverslips back to the parafilmed large Petri dish
- Carefully add 40 µL of the secondary antibody solution to the top of the coverslips. Make sure not to spill the secondary antibody solution over the side of the coverslip.
- If needed, add more ultrapure water to a smaller petri dish and carefully arrange the small petri dish around coverslips.
- Cover the large Petri dish
- For sliced tissue:
- Carefully add 250 µL of the secondary antibody solution to free-floating slices in a 24 well plate.
- Cover the plate
NOTE: From here on, protect samples from light by wrapping the tops of the plates with foil.
- Incubate the samples at 37 °C for 1 h
5. Ligation
- Mix the ligation stock 1:5 in molecular-grade water.
- As in section 4, carefully transfer the coverslips and remove the secondary mix from the sliced tissue.
- Wash the samples in 500 µL of wash buffer A
- For cultured cells, wash 2 times for 5 min. For sliced tissue, wash 2 times for 10 min. Use gentle agitation on an orbital shaker for both.
- During this time, change the parafilm in the large Petri dish
- While keeping the ligase on a cold block, dilute the ligase 1:40 in the ligation stock from step 5.1. Perform this dilution immediately before adding the ligase to the samples.
- As in section 4, remove as much of the wash buffer A as possible from samples before adding the ligase.
- For cultured cells: Transfer coverslips back to the parafilmed Petri dish. Add 40 µL of the ligation mix to coverslips, arrange small water-filled Petri dishes around the coverslips, and cover the large Petri dish.
- For sliced tissue: Add 250 µL of the ligation mix from Step 5.4 to each well and cover the plate.
- Incubate the samples for 30 min at 37 °C.
6. Amplification
- Mix the amplification stock 1:5 in molecular-grade water.
- As in section 4, carefully transfer the coverslips and remove the ligation mix from the sliced tissue.
- Wash samples in 500 µL of wash buffer A
- For cultured cells, wash 2 times for 2 min. For sliced tissue, wash 2 times for 10 min. Use gentle agitation on an orbital shaker for both.
- During this time, change the parafilm in the large Petri dish
- Perform this dilution immediately before adding the polymerase to samples. While keeping the polymerase on a cold block, dilute polymerase
- For cultured cells, dilute polymerase 1:80 in the amplification stock from step 6.1.
- For sliced tissue, dilute polymerase 1:40 in the amplification stock from step 6.1.
- As in step 4, remove as much of the wash buffer A as possible from samples before adding the polymerase.
- For cultured cells: Transfer the coverslips back to the parafilmed Petri dish. Add 40 µL of the amplification mix from Step 6.4.1 to coverslips, arrange small water-filled Petri dishes around the coverslips, and cover the large Petri dish. Incubate the samples for 100 min at 37 °C.
- For sliced tissue: Add 250 µL of the amplification mix from Step 6.4.2 to each well and cover the plate. Incubate the samples for 2 h at 37 °C.
NOTE: During this time, prepare and label slides.
7. Mounting
- As in Step 4, carefully transfer the coverslips and remove the amplification mix from the sliced tissue.
- Wash the samples in 500 µL of wash buffer B 2 times for 10 min with gentle agitation on an orbital shaker.
- Wash the samples in 500 µL of 1% wash buffer B for 1 min with gentle agitation on an orbital shaker.
- For cultured cells:
- Drop 3 µL of mounting media onto a slide
- Tap off excess wash buffer from the coverslip and then place the coverslip (with cells facing down) into the mounting media. Seal the sides with a small amount of clear nail polish to seal the coverslip in place.
- For sliced tissue:
- Carefully transfer a tissue slice to the prepared slide and arrange it, so the slice is lying flat. Drop between 5-10 µL of mounting media (amount will depend on the size of the slice) onto tissue slice
- Carefully place a glass coverslip over tissue slice and seal with a small amount of clear nail polish along the edge to seal the coverslip in place.
- Wait at least 15 min before analyzing under the microscope, or store at -20 °C.
8. Obtain digital images with a confocal microscope
- Optimize the acquisition settings (e.g., laser power, gain, offset) across samples from all treatments. Ensure that the optimization includes decreasing background noise and enhancing the signal without oversaturating the intensity of the fluorescent signals. Once settings are determined, apply the same acquisition settings across all images obtained.
NOTE: The following acquisition details can be used with a Nikon A1 confocal microscope and the Nikon NIS AR Elements software. - Set the slide with sample on the stage and find the focal plane for the sample using DAPI through the eyepiece.
- Turn off the eye port by clicking on Eye port and choose an optical configuration button to adjust the settings.
- Adjust the gain, offset, and laser power for each fluorescent channel to decrease background noise and enhance the fluorescent signal. Make sure the fluorescent signal does not become oversaturated as mentioned in steps 8.5-8.6.
- Monitor oversaturation using a pseudocolor for the fluorescent signal. At the bottom of the live image, right-click the tab labeled with the fluorescent channel currently being used.
- Next, choose Channel Coloring and pick a pseudocolor like Rainbow Dark to visualize the fluorescence intensity in a heat-map-like pseudocolor. In Rainbow Dark, cooler colors indicate less fluorescent intensity, and hotter colors indicate more fluorescent intensity.
- Once all fluorescent channels are optimized, right-click on the optical configuration button previously chosen and choose Assign Current Camera Setting for this button.
- Verify that the chosen settings are sufficient for a random sample from each treatment group. If the chosen settings oversaturate any of these samples, repeat step 8.4 to eliminate the oversaturation.
- For cultured neurons, follow steps 8.10-8.16.
- Using the eye-port, search for a neuron with dendrites that have minimal overlap with other dendrites.
- Turn off the eye port and use the DAPI channel to visualize the cell body of the chosen neuron. Double-click on the center of the soma to center the neuron in the middle of the field of view.
- Using the MAP2 channel, find the best plane of focus for the MAP2 signal with live scanning.
- Under the ND Acquisition tab, click on Save to File and choose a file to save the image into under Parcourir. Then, input the filename.
- Under the Z tab, select the Symmetric Mode Defined by Range option. Set the focus to the best MAP2 plane and click on the Relative button to set this focal plane as the middle of the z-stack.
- Set the range to 5 µm with 1 µm steps, and make sure to check Close Active Shutter During Z Movement. Under the Wavelength tab, select the name of the optical button with the previously configured acquisition settings under Optical Conf. Then, click on Run Now.
- Repeat steps 8.1.10-8.1.15 for about 10 neurons per coverslip/treatment.
- For sliced tissue, follow steps 8.18-8.22.
- Using the eye-port, search for the region of interest. For example, locate CA1 of the hippocampus.
- Turn off the eye-port and use the MAP2 channel to find the best plane of focus for the MAP2 signal with live scanning.
- Under the Acquire menu, choose Scan Large Image. Next, select the optical button's name with the previously configured acquisition settings under the Capturing panel from the panel that opens. Also, ensure to select the correct objective in this panel.
- Under the Area panel and the eyepiece, use the arrow keys to set the boundaries of the region of interest. Next, click on Save Large Image to File and create a save path filename for the image.
- Under the Setup panel, make sure Multichannel Capture is checked, and then choose the name of the optical button with the previously configured acquisition settings under Optical Conf.
NOTE: A z-stack for a large image is possible but will increase the scan time.
9. Analysis
- Similar to acquisition settings, use samples from all treatments to optimize threshold settings. Ensure that the threshold optimization focuses on decreasing background noise and enhancing the signal without oversaturating the intensity of the fluorescent signals. Once these settings are determined, apply the same threshold settings across all images used for analysis as described in steps 9.2-9.3.
- In ImageJ, the threshold option is located under the menu Image > Adjust > Threshold. Choose the Dark Background option if the image has a dark background.
- Next, adjust the upper and lower bounds of the threshold per previously determined optimized threshold settings, then click on Apply.
- For cultured cells, use the MAP2 channel and a freehand region of interest (ROI) tool to draw an ROI for each neuron, including dendrites and soma. For sliced tissue, draw a freehand ROI within the slice image that encapsulates the area of interest (e.g., stratum radiatum of the CA1 within the hippocampus).
- Obtain the area of the ROI. In ImageJ, measure the area under the menu Analyze > Measure.
- Detect the number of puncta within each ROI using an automatic detection tool like Particle Analysis in ImageJ following steps 9.7-9.9.
- Find the Particle Analysis option under the menu Analyze > Analyze Particles. First, define the puncta size diameter, typically 0.1-3 µm2.
- Next, choose the Overlay Masks option from the Show drop-down menu and check the Display Results option. Then, click on OK.
- If puncta are not detected with the diameter range chosen, adjust the range until all puncta are detected with this analysis. Make sure to use the same Particle Analysis settings for all images.
- Divide the number of puncta by the area of an individual region of interest by following steps 9.11-9.13.
- Copy and paste the results for each image from the Results pop-up from ImageJ into a spreadsheet.
- First, identify which file and sample the data were obtained from. Then, divide the area of the ROI by the number of puncta.
- Then, clear the data from the Results pop-up, and repeat steps 9.2-9.12.
- Normalize results to control samples: Average the results (number of puncta/ROI area) for the control samples. Then, divide the obtained results of all samples by the average of the control to obtain the normalized results. The new average of the control samples should be equal to 1.
Data modified from Heaney et al.6 are presented to demonstrate an experiment where increased synapse formation is expected (please see6 for more information and a more in-depth discussion of the mechanism). Previously, it was demonstrated that rapid antidepressants require activation of the inhibitory metabotropic receptor, GABAB (gamma-aminobutyric acid subtype B), to be effective13. Further, previous data indicated that rapid antidepressants increase postsynaptic markers14; thus, DetectSyn was used to test the hypothesis that rapid antidepressants increase synapse numbers to be effective.
In cultured neurons, four conditions are tested. First, in the top panel of Figure 2A, cultured neurons were treated with the vehicle under the same conditions as the experimental control. However, the PSD95 primary antibody is omitted to provide a technical control for the DetectSyn assay (see Figure 1 for how each component contributes to the signal). Next, in the Control panel, neurons again are treated with the vehicle but receive both PSD95 and Synapsin1 primaries. Next, cultured neurons are treated only with the rapid antidepressant Ro-25-6981 (Ro) or Ro plus the GABAB agonist, baclofen (Bac). As previously demonstrated13, both the rapid antidepressant Ro-25-6981 and the GABAB agonist baclofen are required for the in vitro efficacy of Ro-25-6981. Thus, an increase in synapse formation is demonstrated by an increase in white puncta (Figure 2A). Without baclofen, no increase in synapse formation in vitro is seen.
In vivo, basal GABA levels are sufficient for the rapid antidepressant to work13, so only the rapid antidepressant Ro-25-6981 is administered via intraperitoneal injection to mice (Figure 2B). The left panel of Fig. 2B represents another technical control for the DetectSyn assay. Hippocampal tissue from a saline-treated mouse was probed with both primaries for PSD95 and Synapsin1, but one of the secondaries was omitted. An increase in synapse formation due to treatment with the rapid antidepressant Ro-25-6981 compared to saline-treated mice is demonstrated in the middle and right panels of Figure 2B.
Representative images for technical controls in vitro and ex vivo are shown. In Figure 2A, one of the primaries for the synaptic pairs is omitted (i.e., PSD95), and in Figure 2B, one of the secondaries is omitted. While some puncta appear in the technical control images, they are generally not the same size, as demonstrated by the white speck in the top panel of Figure 2A compared to the large puncta in the other panels. Further, these puncta are not in the same location as the puncta quantified, as demonstrated by some puncta appearing within the soma of the technical control in Figure 2B. Typically, non-specific puncta occur within the nuclei, perhaps due to the presence of DNA.
To analyze the representative results in Figure 2A, dendrites (visualized by MAP2 staining and represented here in a grey outline) were traced using a freehand drawing tool to create regions of interest (ROIs). First, the area of the MAP2 ROIs was obtained using the NIS Elements function, ROI Data under the Automated Measurement Results tab. Next, puncta were detected using the Thresholding function in NIS Elements. This function creates a binary mask of objects that fall within a defined intensity threshold. Then, the number of binary objects within the MAP2 ROIs were detected using the NIS Elements function, Binary in ROI, under the Automated Measurement Results tab. The data presented in the graph to the right of Figure 2A are the normalized values of the puncta divided by the area of the ROI.
To analyze the representative results in Figure 2B, the stratum radiatum of the CA1 was traced using a freehand drawing tool to create an ROI. First, the area of the ROI was obtained using the ROI data function, as described in the paragraph above. Next, puncta were detected using the Spot Detection – Bright Spots function, located under the Binary menu. Next, the diameter and contrast values were chosen based on visual inspection of multiple slices from all treatments. Once these values were decided, they were applied uniformly across all analyzed images. The Spot Detection function creates binary masks of objects within the defined diameter and contrast parameters set. Then, the number of binary objects within the MAP2 ROIs were detected using the NIS Elements function, Binary in ROI, under the Automated Measurement Results tab. The data presented in the graph to the right of Figure 2B are the normalized values of the puncta divided by the area of the ROI.
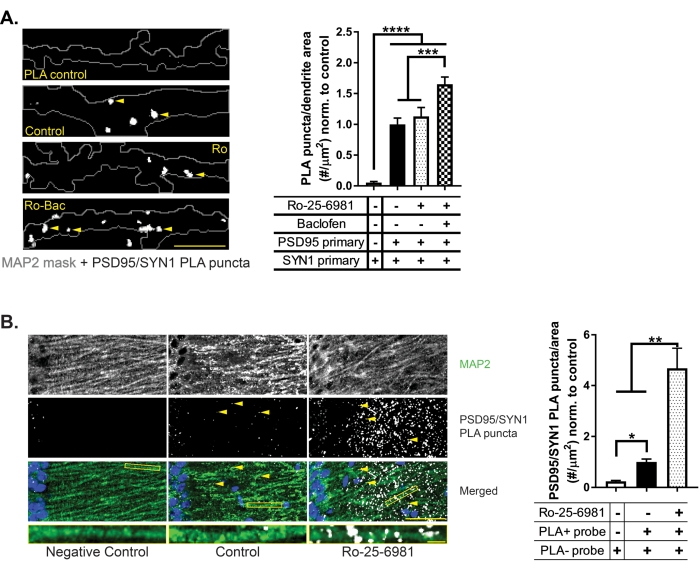
Figure 2: Detection of synapses using DetectSyn assay. White puncta represent synapses detected with DetectSyn PSD95-Synapsin1 proximity ligation assay (yellow arrowheads) in (A) in vitro cultured primary hippocampal neurons and (B) ex vivo CA1 stratum radiatum. In (A), the grey outline represents MAP2 staining. In (A), the top panel labeled PLA control represents samples treated with only vehicle, like the Control. However, the PSD95 primary was not included in the reaction. In the final two panels, neurons were treated with the rapid antidepressant Ro-25-6981 (Ro, 10 µM) or Ro plus the GABAB agonist baclofen (Bac, 50 µM) for 90 min. The increased number of puncta (identified with yellow arrowheads) indicates that in vitro, synapse formation only increases when the rapid antidepressant Ro-25-6981 is administered with the GABAB agonist. In (B), green represents dendrites stained with MAP2, and blue represents nuclei stained with DAPI. Single dendrites, outlined in yellow rectangles in the merged images, are isolated beneath representative images to demonstrate that puncta localize to dendrites. Animals were treated with Ro-25-6981 (10 mg/kg), and tissue was collected 45 min after treatment. The increased number of puncta (identified by yellow arrowheads) indicates that in vivo, the number of synapses increases with basal GABA signaling and the rapid antidepressant Ro-25-6981. Quantification of representative results is represented by the bar graphs and indicates the average number of DetectSyn puncta divided by MAP2 area. Results are normalized to the experimental control. Images were obtained using a Nikon A1plus confocal microscope. Scale bars = (A) 10 µm; (B, top) 50 µm; (B, bottom) 5 µm. Bars represent the mean +/- standard error of the mean. Results analyzed by one-way ANOVA: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 by post-hoc analysis. The figure has been modified from Heaney et al.6. Please click here to view a larger version of this figure.
