Notre compréhension de la différenciation cellulaire et de la genèse des tissus et des organes est le résultat de décennies de criblages ciblés élaborés de gènes et de leurs produits. Accroître notre connaissance de toutes les biomolécules et de leurs quantités lors d’événements cellulaires importants aiderait à démêler les mécanismes moléculaires qui contrôlent la structuration spatiale et temporelle du plan corporel des vertébrés. Les technologies permettant l’amplification moléculaire et le séquençage sont maintenant en mesure de rendre compte régulièrement d’un grand nombre de gènes et de transcrits à l’appui d’études fondées sur des hypothèses dans la recherche biologique fondamentale et translationnelle. Pour comprendre les systèmes en développement, une relation complexe entre la transcription et la traduction plaide en faveur de l’analyse directe de multiples protéines et de leurs modifications post-traductionnelles. La protéomique globale utilisant des systèmes biologiques in vitro, tels que les cellules souches pluripotentes induites, a commencé à délimiter les mécanismes d’induction tissulaire 1,2. Dans les organismes complexes, tels que l’embryon de vertébrés, le développement repose sur les gradients morphogènes dans le contexte de l’espace et du temps3. Il s’ensuit que l’acquisition de connaissances sur les changements protéomiques à mesure que les cellules se différencient pour former des tissus spécialisés, tels que les tissus neuraux, offre une clé pour débloquer des programmes moléculaires contrôlant le développement normal et défectueux et guider les thérapies de prochaine génération.
La grenouille à griffes vertébrée d’Afrique du Sud (Xenopus laevis) est un modèle bien établi en biologie cellulaire et développementale, neuro- et régénérative. Le prix Nobel de physiologie ou médecine 4,5 de Sir John Gurdon en 2012 pour la découverte de la pluripotence du noyau somatique a souligné l’importance de ce modèle pour les découvertes en études fondamentales et translationnelles. Les embryons Xenopus se développent à l’extérieur de la mère, facilitant ainsi la manipulation directe des cellules, des clones cellulaires et de l’expression génique à différents stades de développement. La pigmentation asymétrique et les divisions cellulaires stéréotypées ont permis de cartographier des cartes de devenir reproductibles à partir de l’embryon de stade 7,8 à 16-6 et 32 cellules. Pour la protéomique basée sur la spectrométrie de masse à haute résolution (SGRH), les avantages supplémentaires du modèle comprennent une taille relativement grande (~1 mm de diamètre), qui donne une teneur abondante en protéines pour l’analyse (~130 μg chez les embryons au stade de clivage précoce, ~10 μg de teneur en protéines dans les cellules individuelles de l’embryon à 16 cellules)9,10.
À l’heure actuelle, le SGRH est la principale technologie de choix pour la détection des protéines. Cette technologie permet la détection et la quantification directes, sensibles et spécifiques de multiples, généralement des centaines, voire des milliers de protéines différentes11. La protéomique ascendante du SGRH comporte une série d’étapes interconnectées. Après l’extraction de l’échantillon de cellule/tissu, les protéines sont digérées avec une enzyme protéolytique, telle que la trypsine (protéomique ascendante). Les peptides résultants sont séparés en fonction de leurs différentes propriétés physico-chimiques, y compris l’hydrophobicité (chromatographie liquide en phase inverse, LC), la charge nette (chromatographie par échange d’ions), la taille (chromatographie d’exclusion de taille) ou la mobilité électrophorétique (électrophorèse capillaire, CE). Les peptides sont ensuite chargés (ionisés), généralement à l’aide d’une ionisation par électronébulisation (ESI), et les ions peptidiques sont détectés et séquencés par fragmentation en phase gazeuse par HRMS en tandem. Les données peptidiques résultantes sont mises en correspondance avec le protéome de l’organisme étudié. Avec l’intensité du signal d’ions peptidiques spécifiques aux protéines (protéotypiques) corrélée à la concentration, la quantification des protéines peut être effectuée sans marquage ou sur la base du marquage (quantification multiplexage). La protéomique du SGRH fournit une riche source d’information sur l’état moléculaire du système à l’étude, ce qui permet de générer des hypothèses et des études fonctionnelles de suivi.
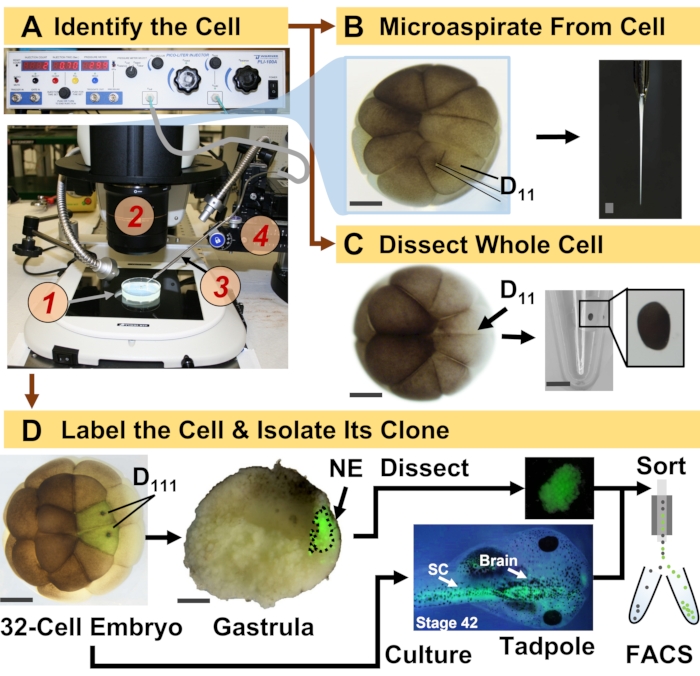
Figure 1 : Protéomique spatio-temporellement évolutive permettant la protéomique HRMS guidée par lignée cellulaire dans l’embryon (grenouille) en développement. (A) Visualisation de l’échantillon (1) à l’aide d’un stéréomicroscope (2) pour l’injection d’une cellule identifiée (encadré), à l’aide d’une micropipette fabriquée (3) sous contrôle par un étage de translation (4). (B) Prélèvement subcellulaire de la cellule D 11 gauche identifiée dans un embryon à16 cellules. (C) Dissection d’une cellule D 11 entière à partir d’un embryon à16 cellules. (D) Traçage fluorescent (vert) des descendants D111 gauche et droit d’un embryon à 32 cellules pour guider la dissection de l’ectoderme neural (NE) dans la gastrule (stade 10) et isolement du tissu descendant du têtard à l’aide de FACS. Barres d’échelle : 200 μm pour les embryons, 1,25 mm pour le flacon. Les figures ont été adaptées avec la permission des références 15,19,21,59. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Le protocole présenté ici permet de quantifier un grand nombre de protéines dans des cellules/tissus identifiés dans des embryons de X. laevis en développement basés sur le SGRH. L’approche s’appuie sur une identification cellulaire précise, des cartes de destin cellulaire reproductibles et des méthodologies établies pour suivre les lignées cellulaires dans ce modèle biologique 6,7,8. Comme le montre la figure 1, nous étudions les protéomes de cellules individuelles en utilisant la dissection de cellules entières ou le microéchantillonnage capillaire pour aspirer le contenu cellulaire. Le suivi de la lignée d’une cellule nous permet d’étudier l’évolution spatio-temporelle du protéome lorsque les cellules forment des tissus pendant la gastrulation. La descendance cellulaire est marquée par fluorescence par injection d’un fluorophore conjugué à du dextrane inerte ou à l’ARNm pour la protéine fluorescente (p. ex. protéine fluorescente verte, ou GFP). La descendance marquée est isolée aux moments de développement souhaités. Pendant la gastrulation, les clones cellulaires étroitement regroupés peuvent être isolés par dissection. Après gastrulation, les clones cellulaires peuvent être distribués dans l’embryon en raison des mouvements migratoires et peuvent être isolés des tissus dissociés par tri cellulaire activé par fluorescence (FACS). Les protéines de ces cellules et tissus sont mesurées par protéomique ascendante utilisant HPLC ou CE pour la séparation et ESI tandem HRMS pour l’identification. La protéomique HRMS guidée par lignée cellulaire est évolutive à différentes tailles de cellules et lignées au sein de l’embryon et est spécifique, sensible et quantitative. À travers des exemples sélectionnés présentés ici, nous démontrons également que ce protocole est évolutif et largement adaptable à différents types de cellules et de lignées cellulaires.
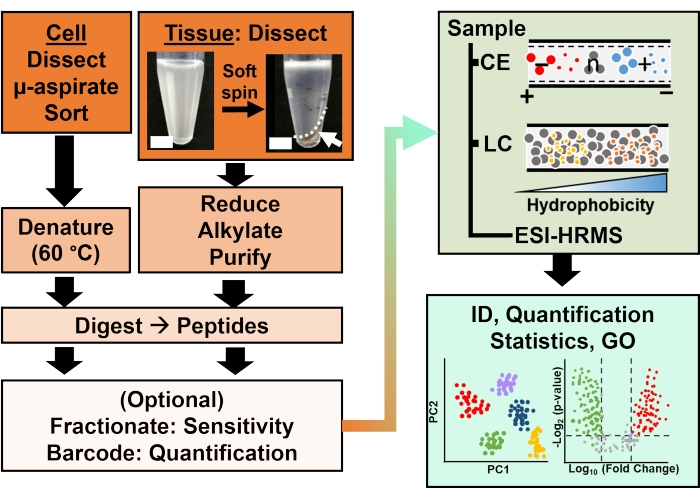
Figure 2 : Le flux de travail bioanalytique. La micro-dissection et l’aspiration capillaire, ou FACS, ont facilité l’échantillonnage de la teneur en protéines cellulaires et clonales. Épuisement des protéines vitellines abondantes et séparation par électrophorèse capillaire (EC) ou chromatographie liquide à nanoflux (LC) sensibilité à l’identification améliorée (ID) à l’aide de la spectrométrie de masse à haute résolution (HRMS) par ionisation électronébulaire (ESI). La quantification a révélé une dysrégulation, fournissant de nouvelles informations pour des études basées sur des hypothèses en conjonction avec les informations disponibles à partir de l’ontologie génique (GO). Les chiffres ont été adaptés avec la permission de la référence15. Veuillez cliquer ici pour voir une version agrandie de cette figure.
