Vår forståelse av celledifferensiering og opprinnelsen til vev og organer er resultatet av flere tiår med forseggjorte målrettede skjermer av gener og deres produkter. Å øke vår kunnskap om alle biomolekylene og deres mengder under viktige cellulære hendelser vil bidra til å avdekke molekylære mekanismer som styrer romlig og tidsmessig mønster av virveldyrets kroppsplan. Teknologier som muliggjør molekylær forsterkning og sekvensering er nå i stand til rutinemessig å rapportere om et stort antall gener og transkripsjoner, og støtter hypotesedrevne studier i grunnleggende biologisk og translasjonsforskning. For å forstå utvikling av systemer, tar et komplekst forhold mellom transkripsjon og oversettelse til orde for direkte analyse av flere proteiner og deres posttranslasjonelle modifikasjoner. Global proteomikk ved bruk av in vitro biologiske systemer, slik som induserte pluripotente stamceller, begynte å avgrense mekanismer for vevsinduksjon 1,2. I komplekse organismer, som virveldyrembryoet, er utviklingen avhengig av morfogengradienter i sammenheng med rom og tid3. Det følger at å få kunnskap om proteomiske forandringer når celler skiller seg for å danne spesialiserte vev, for eksempel nevrale vev, gir en nøkkel til å låse opp molekylære programmer som kontrollerer normal og defekt utvikling og veilede neste generasjons terapi.
Virveldyret sørafrikansk klørfrosk (Xenopus laevis) er en veletablert modell innen celle- og utviklings-, nevro- og regenerativ biologi. Sir John Gurdons 2012 Nobelpris i fysiologi eller medisin 4,5 for oppdagelsen av pluripotens av den somatiske kjernen fremhevet betydningen av denne modellen for funn i grunnleggende og translasjonsstudier. Xenopus-embryoer utvikler seg eksternt til moren, og letter dermed direkte manipulering av celler, cellekloner og genuttrykk over ulike utviklingsstadier. Asymmetrisk pigmentering og stereotype celledelinger gjorde det mulig å kartlegge reproduserbare skjebnekart fra 16-6 og 32-celle 7,8 stadium embryo. For høyoppløselig massespektrometri (HRMS) basert proteomikk inkluderer ytterligere fordeler med modellen relativt stor størrelse (~ 1 mm i diameter), noe som gir rikelig proteininnhold for analyse (~ 130 μg i tidlige spaltningsstadiumembryoer, ~ 10 μg proteininnhold i enkeltceller i 16-celleembryoet) 9,10.
For tiden er HRMS den ledende teknologien som er valgt for å oppdage proteiner. Denne teknologien muliggjør direkte, sensitiv og spesifikk deteksjon og kvantifisering av flere, vanligvis hundrevis-til-tusenvis av forskjellige proteiner11. Bottom-up proteomikk av HRMS innebærer en rekke sammenkoblede trinn. Etter ekstraksjon fra celle-/vevsprøven fordøyes proteiner med et proteolytisk enzym, slik som trypsin (bottom-up proteomics). De resulterende peptidene separeres basert på deres forskjellige fysisk-kjemiske egenskaper, inkludert hydrofobicitet (reversert fasevæskekromatografi, LC), nettoladning (ionebytterkromatografi), størrelse (størrelsesekskluderingskromatografi) eller elektroforetisk mobilitet (kapillær elektroforese, CE). Peptider blir deretter ladet (ionisert), vanligvis ved bruk av elektrosprayionisering (ESI), og peptidioner detekteres og sekvenseres via gassfasefragmentering av tandem-HRMS. De resulterende peptiddataene kartlegges til proteomet til organismen som studeres. Med proteinspesifikk (proteotypisk) peptidionsignalintensitet korrelert med konsentrasjon, kan proteinkvantifisering utføres etikettfri eller etikettbasert (multipleksingskvantifisering). HRMS proteomikk gir en rik ressurs av informasjon om molekylær tilstand av systemet under studien, noe som gjør det mulig å generere hypoteser og oppfølging funksjonelle studier.
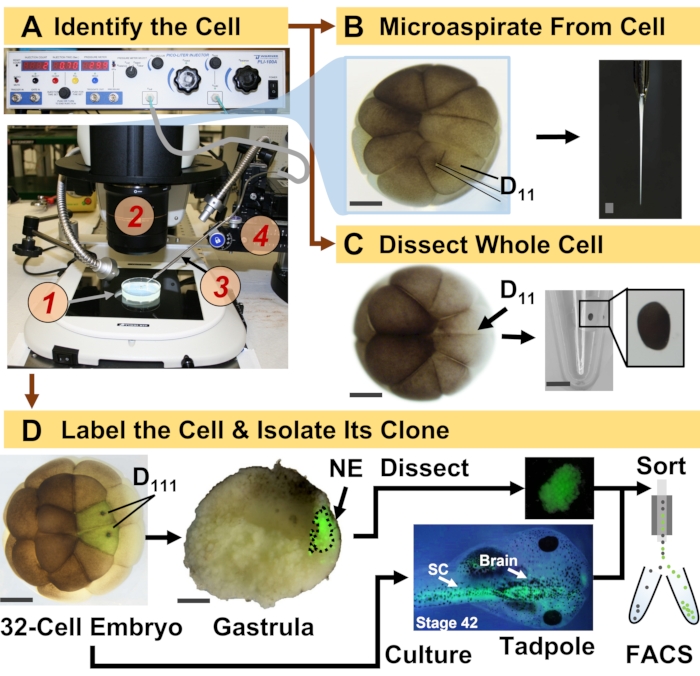
Figur 1: Spatiotemporalt skalerbar proteomikk som muliggjør cellelinjeveiledet HRMS-proteomikk i det utviklende (froske) embryoet. (A) Visualisering av prøven (1) ved hjelp av et stereomikroskop (2) for injeksjon av en identifisert celle (innfelt), ved bruk av en fabrikert mikropipette (3) under kontroll av et oversettelsestrinn (4). (B) Subcellulær prøvetaking av den identifiserte venstre D11-cellen i et 16-celleembryo. (C) Disseksjon av en hel D11 celle fra et 16-celle embryo. (D) Fluorescerende (grønn) sporing av venstre og høyre D111 progenier fra et 32-celleembryo for å lede disseksjon av nevralektoderm (NE) i gastrula (trinn 10) og isolering av etterkommervevet fra rumpetroll ved hjelp av FACS. Skalastenger: 200 μm for embryoer, 1,25 mm for hetteglasset. Tallene ble tilpasset med tillatelse fra referansene 15,19,21,59. Klikk her for å se en større versjon av denne figuren.
Protokollen som presenteres her muliggjør HRMS-basert kvantifisering av et stort antall proteiner i identifiserte celler/vev i utviklingen av X. laevis-embryoer. Tilnærmingen bygger på nøyaktig celleidentifikasjon, reproduserbare celleskjebnekart og etablerte metoder for å spore cellelinjer i denne biologiske modellen 6,7,8. Som vist i figur 1 studerer vi proteomer fra enkeltceller ved å bruke helcelledisseksjon eller kapillær mikrosampling for å aspirere cellulært innhold. Overvåking av avstamningen til en celle tillater oss å studere den spatiotemporale utviklingen av proteomet når celler danner vev under gastrulasjon. Celleavkommet er fluorescerende merket ved å injisere en fluorofor konjugert til inert dextran eller mRNA for fluorescerende protein (f.eks. grønt fluorescerende protein eller GFP). Det merkede avkommet isoleres ved ønskede utviklingstidspunkter. Under gastrulering kan cellekloner som er tett gruppert isoleres ved disseksjon. Etter gastrulering kan cellekloner distribueres i embryoet på grunn av migrerende bevegelser og kan isoleres fra dissosierte vev ved fluorescensaktivert cellesortering (FACS). Proteiner i disse cellene og vevene måles via bottom-up-proteomikk som bruker HPLC eller CE for separasjon og ESI-tandem HRMS for identifikasjon. Cellelinjestyrt HRMS-proteomikk er skalerbar til forskjellige cellestørrelser og linjer i embryoet og er spesifikk, sensitiv og kvantitativ. Gjennom utvalgte eksempler vist her, demonstrerer vi også at denne protokollen er skalerbar og bredt tilpasningsdyktig til forskjellige typer celler og cellelinjer.
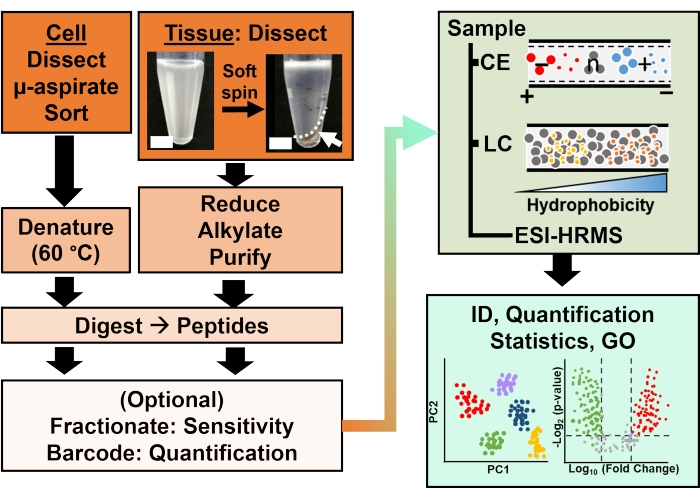
Figur 2: Den bioanalytiske arbeidsflyten. Mikrodisseksjon og kapillær aspirasjon, eller FACS muliggjorde prøvetaking av cellulært og klonalt proteininnhold. Uttømming av rikelig eggeplommeproteiner og separasjon ved kapillær elektroforese (CE) eller nano-flow væskekromatografi (LC) forbedret identifikasjon (ID) følsomhet ved bruk av elektrosprayionisering (ESI) høyoppløselig massespektrometri (HRMS). Kvantifisering avslørte dysregulering, og ga ny informasjon for hypotesedrevne studier i forbindelse med informasjon tilgjengelig fra genontologi (GO). Figurene ble bearbeidet med tillatelse frareferanse 15. Klikk her for å se en større versjon av denne figuren.
