Наше понимание дифференцировки клеток и генезиса тканей и органов является результатом десятилетий сложных целевых скринингов генов и их продуктов. Расширение наших знаний обо всех биомолекулах и их количествах во время важных клеточных событий поможет разгадать молекулярные механизмы, которые контролируют пространственную и временную структуру плана тела позвоночных. Технологии, обеспечивающие молекулярную амплификацию и секвенирование, теперь могут регулярно сообщать о большом количестве генов и транскриптов, поддерживая исследования, основанные на гипотезах, в фундаментальных биологических и трансляционных исследованиях. Чтобы понять развивающиеся системы, сложная взаимосвязь между транскрипцией и трансляцией требует прямого анализа нескольких белков и их посттрансляционных модификаций. Глобальная протеомика с использованием биологических систем in vitro, таких как индуцированные плюрипотентные стволовые клетки, начала очерчивать механизмы тканевой индукции 1,2. У сложных организмов, таких как эмбрион позвоночных, развитие зависит от градиентов морфогена в контексте пространства и времени3. Из этого следует, что получение знаний о протеомных изменениях по мере того, как клетки дифференцируются с образованием специализированных тканей, таких как нервные ткани, дает ключ к разблокированию молекулярных программ, контролирующих нормальное и дефектное развитие, и направляет терапию следующего поколения.
Позвоночная южноафриканская когтистая лягушка (Xenopus laevis) является хорошо зарекомендовавшей себя моделью в клеточной и развивающейся, нейро- и регенеративной биологии. Нобелевская премия сэра Джона Гердона по физиологии и медицине 2012 года 4,5 за открытие плюрипотентности соматического ядра подчеркнула важность этой модели для открытий в фундаментальных и трансляционных исследованиях. Эмбрионы Xenopus развиваются внешне по отношению к матери, тем самым облегчая прямое манипулирование клетками, клеточными клонами и экспрессией генов на различных стадиях развития. Асимметричная пигментация и стереотипное деление клеток позволили составить карту воспроизводимых карт судьбы 16-6 и 32-клеточногоэмбриона на 7,8 стадии. Для протеомики на основе масс-спектрометрии высокого разрешения (HRMS) дополнительные преимущества модели включают относительно большой размер (~1 мм в диаметре), что дает обильное содержание белка для анализа (~130 мкг у эмбрионов на ранней стадии расщепления, ~10 мкг белка в одиночных клетках 16-клеточного эмбриона)9,10.
В настоящее время HRMS является ведущей технологией выбора для обнаружения белков. Эта технология позволяет прямо, чувствительно и специфически обнаруживать и количественно определять множественные, обычно от сотен до тысяч различныхбелков11. Восходящая протеомика HRMS включает в себя ряд взаимосвязанных этапов. После экстракции из образца клетки / ткани белки перевариваются протеолитическим ферментом, таким как трипсин (восходящая протеомика). Полученные пептиды разделяют на основе их различных физико-химических свойств, включая гидрофобность (обращенно-фазовая жидкостная хроматография, LC), суммарный заряд (ионообменная хроматография), размер (эксклюзионная хроматография) или электрофоретическую подвижность (капиллярный электрофорез, CE). Затем пептиды заряжаются (ионизируются), как правило, с помощью электрораспыления ионизации (ESI), а пептидные ионы обнаруживаются и секвенируются посредством газофазной фрагментации с помощью тандемной HRMS. Полученные пептидные данные сопоставляются с протеомом исследуемого организма. При том, что интенсивность сигнала пептид-ионов специфического белка (протеотипа) коррелирует с концентрацией, количественное определение белка может быть выполнено без меток или на основе меток (мультиплексирующее количественное определение). Протеомика HRMS дает богатый источник информации о молекулярном состоянии исследуемой системы, что позволяет генерировать гипотезы и проводить последующие функциональные исследования.
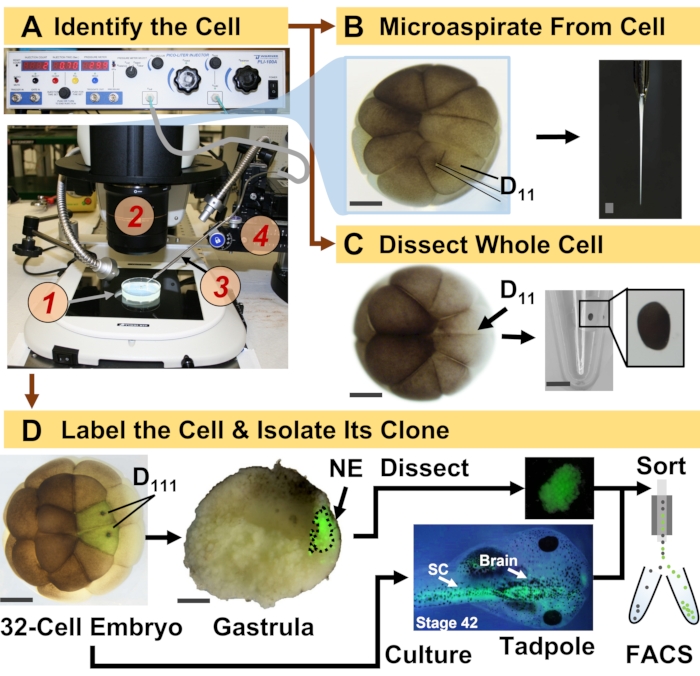
Рисунок 1: Пространственно-временная масштабируемая протеомика, обеспечивающая протеомику HRMS, управляемую клеточной линией, в развивающемся эмбрионе (лягушке). (A) Визуализация образца (1) с использованием стереомикроскопа (2) для инъекции идентифицированной клетки (врезка) с использованием изготовленной микропипетки (3) под контролем трансляционной стадии (4). (B) Субклеточный отбор идентифицированных левых клеток D11 в 16-клеточном эмбрионе. (C) Рассечение целой клетки D11 из 16-клеточного эмбриона. (D) Флуоресцентное (зеленое) отслеживание левого и правого потомства D111 от 32-клеточного эмбриона для направления рассечения невральной эктодермы (NE) в гаструле (стадия 10) и выделения ткани-потомка от головастика с использованием FACS. Масштабные линейки: 200 мкм для эмбрионов, 1,25 мм для флакона. Рисунки были адаптированы с разрешения ссылок 15,19,21,59. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Представленный здесь протокол позволяет количественно оценить большое количество белков в идентифицированных клетках/тканях развивающихся эмбрионов X. laevis на основе HRMS. Подход основан на точной идентификации клеток, воспроизводимых картах клеточной судьбы и установленных методологиях отслеживания клеточных линий в этой биологической модели 6,7,8. Как показано на рисунке 1, мы изучаем протеомы из отдельных клеток, используя диссекцию целых клеток или капиллярный микропробоотбор для аспирации клеточного содержимого. Наблюдение за происхождением клетки позволяет нам изучать пространственно-временную эволюцию протеома по мере того, как клетки образуют ткани во время гаструляции. Клеточное потомство флуоресцентно маркируют путем введения флуорофора, конъюгированного с инертным декстраном или мРНК для флуоресцентного белка (например, зеленого флуоресцентного белка или GFP). Меченое потомство изолируют в желаемые моменты развития. Во время гаструляции клоны клеток, которые плотно сгруппированы, могут быть выделены путем рассечения. После гаструляции клеточные клоны могут быть распределены внутри эмбриона за счет миграционных движений и могут быть выделены из диссоциированных тканей с помощью флуоресцентно-активированной сортировки клеток (FACS). Белки в этих клетках и тканях измеряются с помощью восходящей протеомики с использованием ВЭЖХ или КЭ для разделения и тандемной HRMS ESI для идентификации. Протеомика HRMS, управляемая клеточными линиями, масштабируется для различных размеров клеток и линий внутри эмбриона и является специфической, чувствительной и количественной. На отдельных примерах, показанных здесь, мы также демонстрируем, что этот протокол является масштабируемым и широко адаптируемым к различным типам клеток и клеточных линий.
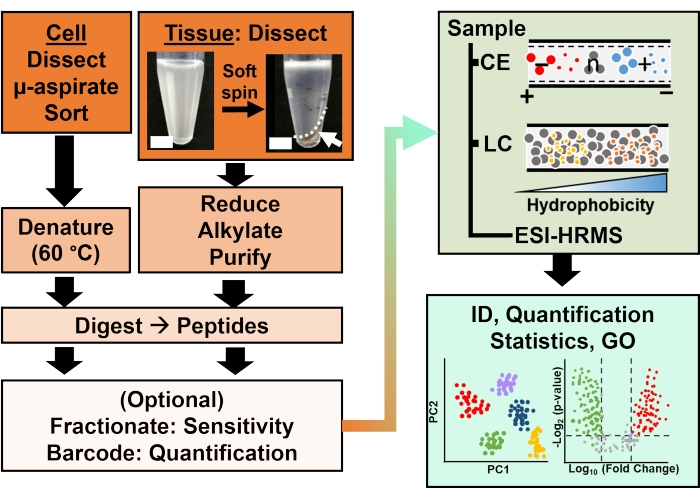
Рисунок 2: Биоаналитический рабочий процесс. Микродиссекция и капиллярная аспирация, или FACS, облегчают отбор проб клеточного и клонального белка. Истощение обильных белков желтка и разделение с помощью капиллярного электрофореза (CE) или нанопоточной жидкостной хроматографии (LC) с повышенной чувствительностью идентификации (ID) с использованием масс-спектрометрии высокого разрешения (HRMS) с помощью ионизации электрораспылением (ESI). Количественная оценка выявила дисрегуляцию, предоставив новую информацию для исследований, основанных на гипотезах, в сочетании с информацией, доступной из онтологии генов (GO). Рисунки были адаптированы с разрешения ссылки15. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
