Tooth organoid development
We provide a detailed protocol to establish organoid cultures from human DF tissue acquired following wisdom tooth extraction (Figure 1A). Isolated DF is enzymatically and mechanically dissociated. The obtained cells are cultured within BMM in media that were empirically defined for optimal organoid development and growth (tooth organoid medium; TOM)19.
The organoids typically develop within 2 weeks after DF cell seeding (P0; Figure 2A). The organoids are long-term expandable (up to 11 passages so far) (Figure 2B, shown at P4). Seeding around 20,000 cells per BMM droplet (at both P0 and further passages) yields an optimal density of organoids (Figure 2C), whereas seeding higher cell numbers leads to suboptimal organoid outgrowth (i.e., smaller organoids at too high density) as there is insufficient space to grow (Figure 2D). The eventually optimized culture conditions allow the development of organoids from DF samples at 100% efficiency19.
Tooth organoid characterization and validation
The developed organoids show a dense appearance and contain cells displaying a high nucleo-cytoplasmic ratio, as similarly observed in ERM cells7 (Figure 3A). Moreover, and in further analogy, the organoids express the ERM marker cytokeratin 14 (CK14)22, thereby confirming their epithelial origin (Figure 3B), as well as other proposed ERM markers (such as P63, CD44 and ITGα612,22,23 (Figure 3B). Moreover, organoids express SOX2, a well-known DESC marker in mice and also present in the epithelium of developing human teeth (Figure 3B)1. Interestingly, amelogenin (AMELX), the main component of the enamel matrix, also found expressed in the organoids, is also detected in the ERM24 (Figure 3C). Expression of still other ERM/stemness markers is described in our recent study19 and can be used to further certify the obtained organoids. Furthermore, the organoids retain their ERM/stemness phenotype during passaging, among others shown by stable expression of ERM/stem cell markers (Figure 3D). Finally, the tooth-derived organoids show differentiation capacity to ameloblast(-like) cells, which can also be applied to validate the obtained organoid cultures, showing expression of mature ameloblast markers such as odontogenic-ameloblast associated protein (ODAM) and amelotin (AMTN) after transfer to differentiation medium (see19).
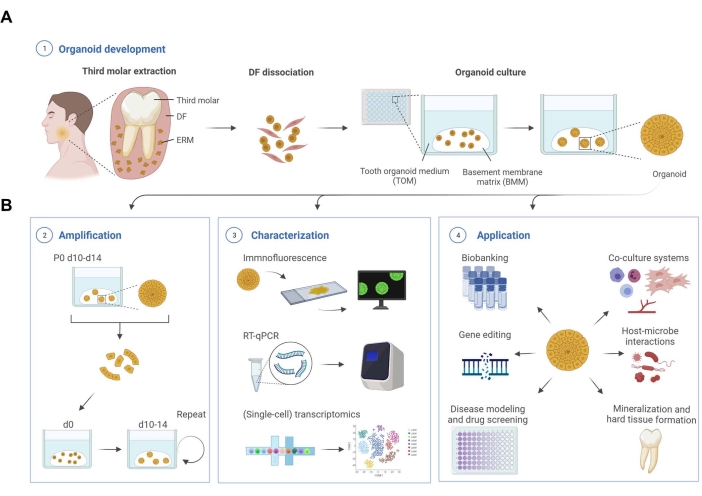
Figure 1: Schematic workflow of tooth organoid development, characterization, and applications. (A) Tooth organoid development from dental follicle (DF) tissue isolated from unerupted third molars. (B) Amplification, characterization, and application potential of tooth organoids. d, day; P, passage. Created with BioRender.com. Please click here to view a larger version of this figure.
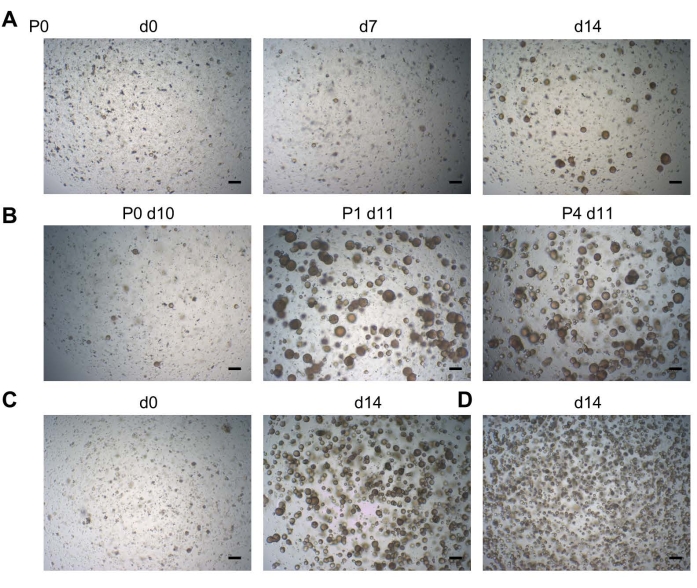
Figure 2: Tooth organoid development. (A) Organoid development from DF tissue. Representative brightfield images shown at different days (d) after seeding (P0; P, passage; 2.5x). (B) Brightfield images showing robust passageability of a tooth organoid line (2.5x). (C) Brightfield images showing a tooth organoid line immediately at passaging (d0; left; 2.5x) seeded at a density of 20,000 cells per well, and the resultant confluent organoid culture ready to be passaged (d14; right; 2.5x). (D) Brightfield images showing a tooth organoid line, which had been seeded at a density of >20,000 cells, leading to smaller organoids at too high density at d14 (2.5x). Scale bars: 200 µm. Please click here to view a larger version of this figure.
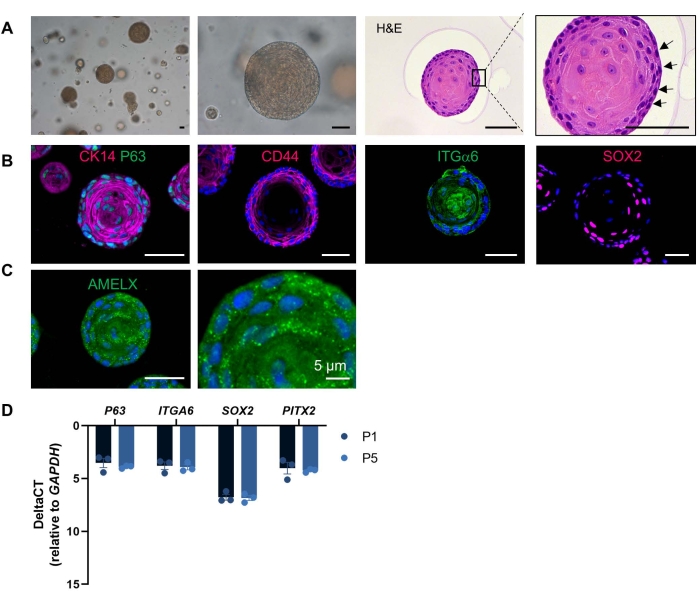
Figure 3: Tooth organoid characterization. (A) Brightfield images of DF-derived organoid cultures at different magnifications showing dense structures developed at 14 days in TOM (P4; 5-20x)). Hematoxylin and eosin staining of an organoid (P1, day 11). Box is enlarged. Arrows indicate cells with a high nucleo-cytoplasmic ratio. (B) Immunofluorescent staining for epithelial/ERM/stemness markers in TOM-grown organoids (20x). (C) Immunofluorescent staining for amelogenin (AMELX) in TOM-grown organoid (20x). DAPI (blue) was used to label nuclei. (D) Gene expression levels (relative to GAPDH) of ERM/stemness markers in P1 and P5 TOM-grown organoids at day 14 of culture (mean ± SEM; n = 3 biological replicates). Scale bars: 50 µm, unless indicated otherwise. Please click here to view a larger version of this figure.
| Dental follicle (DF) collection medium | |
| Name | Concentration |
| Minimum essential medium eagle (αMEM) | |
| Fetal bovine serum (FBS) | 10% |
| Amphotericin B | 0.5% |
| Penicillin-streptomycin (Pen/Strep) | 1% |
Table 1: Dental follicle (DF) collection medium. The table lists the constituents of the DF collection medium.
| Tooth organoid medium (TOM) | |
| Name | Concentration |
| Serum-free defined medium (SFDM) | See Table 3 for composition |
| A83-01 | 0.5 µM |
| B27 (without vitamin A) | 2% |
| Cholera Toxin | 100 ng/mL |
| FGF2 (= basic FGF) | 20 ng/mL |
| FGF8 | 200 ng/mL |
| FGF10 | 100 ng/mL |
| L-Glutamine | 2 mM |
| IGF-1 | 100 ng/mL |
| N2 | 1% |
| N-acetyl L-cysteine | 1.25 mM |
| Nicotinamide | 10 mM |
| Noggin | 100 ng/mL |
| RSPO1 | 200 ng/mL |
| SB202190 (p38i) | 10 µM |
| SHH | 100 ng/mL |
| WNT3a | 200 ng/mL |
Table 2: Tooth organoid medium (TOM). The table lists the constituents and their respective concentrations required to prepare the tooth organoid medium.
| Serum-free defined medium (SFDM) (pH 7.3) | |
| Name | Concentration |
| Sterile H2O | |
| DMEM 1:1 F12 without Fe | 16.8 g/L |
| Transferrin | 5 mg/L |
| Insulin from bovine pancreas | 5 mg/L |
| Penicillin G sodium salt | 35 mg/L |
| Streptomycin sulfate salt | 50 mg/L |
| Ethanol absolute, ≥99.8% (EtOH) | 600 µL/L |
| Catalase from bovine liver | 50 µL/L |
| Sodium Hydrogen Carbonate (NaHCO3) | 1 g/L |
| Albumin Bovine (cell culture grade) | 5 g/L |
Table 3: Serum-free defined medium (SFDM) (pH 7.3). The table lists the composition of the serum-free defined medium.
| Dissociation medium | |
| Name | Concentration |
| Phosphate buffered saline (PBS) | |
| Collagenase IV | 3 mg/mL |
| Dispase II | 4 mg/mL |
Table 4: Dissociation medium. List of constituents and their required concentrations for preparing the dissociation medium.
| Medium A (pH 7.3) | |
| Name | Concentration |
| Sterile H2O | |
| DMEM powder high glucose | 13.38 g/L |
| HEPES | 5.958 g/L |
| Sodium-pyruvate (C3H3NaO3) | 110 mg/L |
| Penicillin G sodium salt | 35 mg/L |
| Streptomycin sulfate salt | 50 mg/L |
| Sodium Chloride (NaCl) | 0.5 g/L |
| Sodium Hydrogen Carbonate (NaHCO3) | 1 g/L |
| Albumin Bovine (cell culture grade) | 3 g/L |
| Dnase* | 0.2 mg/mL |
| *add when mentioned |
Table 5: Medium A (pH 7.3). The table lists the concentration of the constituents used to prepare Medium A.
| Primary Antibodies | ||
| Name | Host | Concentration |
| AMELX | mouse | 1:100 |
| CD44 | mouse | 1:200 |
| CK14 | mouse | 1:200 |
| ITGA6 | rabbit | 1:200 |
| P63 | rabbit | 1:1000 |
| SOX2 | rabbit | 1:2000 |
| Secundary Antibodies | ||
| Name | Host | Concentration |
| mouse IgG (Alexa 555) | donkey | 1:1000 |
| rabbit IgG (Alexa 488) | donkey | 1:1000 |
Table 6: List of antibodies and their dilutions. The table lists the antibodies and their respective dilutions used in this study.
| Primers | ||
| Gene | Forward primer | Reverse primer |
| GAPDH | GGTATCGTGGAAGGACTCATGAC | ATGCCAGTGAGCTTCCCGTTCAG |
| P63 | CAACGCAGTAGACACCATTTCC | CCCAAAACCTTCTCGCTTGTT |
| ITGA6 | GGCGGTGTTATGTCCTGAGTC | AATCGCCCATCACAAAAGCTC |
| SOX2 | GCTGGGACATGTGAAGTCTG | CCCTGTGGTTACCTCTTCCT |
| PITX2 | CAGCGGACTCACTTTACCAG | ATTCTTGAACCAAACCCGGAC |
Table 7: List of primers. The table lists the primers of GAPDH, P63, ITGA6, SOX2, and PITX2 used in this study.
