Isolation, Culture, and Characterization of Primary Schwann Cells, Keratinocytes, and Fibroblasts from Human Foreskin
Summary
The study of wound healing associated with musculoskeletal injury often requires the assessment of in vitro interactions between Schwann cells (SCs), keratinocytes, and fibroblasts. This protocol describes the isolation, culturing, and characterization of these primary cells from the human foreskin.
Abstract
This protocol describes isolation methods, culturing conditions, and characterization of human primary cells with high yield and viability using rapid enzymatic dissociation of skin. Primary keratinocytes, fibroblasts, and Schwann cells are all harvested from the human newborn foreskin, which is available following standard of care procedures. The removed skin is disinfected, and the subcutaneous fat and muscle are removed using a scalpel. The method consists of enzymatic and mechanical separation of epidermal and dermal layers, followed by additional enzymatic digestion to obtain single-cell suspensions from each of these skin layers. Finally, single cells are grown in appropriate cell culture media following standard cell culture protocols to maintain growth and viability over weeks. Together, this simple protocol allows isolation, culturing, and characterization of all three cell types from a single piece of skin for in vitro evaluation of skin-nerve models. Additionally, these cells can be used together in co-cultures to gauge their effects on each other and their responses to in vitro trauma in the form of scratches performed robotically in the culture associated with wound healing.
Introduction
Primary cells derived from living tissue and cultured under in vitro conditions closely resemble the physiological state1, making them an ideal model for investigating physiological and pathophysiological processes. The skin contains multiple cell types, including keratinocytes, fibroblasts, sebocytes, melanocytes, and Schwann cells (SCs), which can be isolated and cultured for in vitro experiments. Methods to isolate and culture keratinocytes, fibroblasts, and SCs, from a single piece of skin, have not been described. The goal of this protocol is twofold: 1) to establish a reliable and reproducible method for the isolation of and culturing of dermal SCs and 2) to use an efficient, robust method for the isolation of keratinocytes, fibroblasts, and SCs from a single human foreskin.
At present, there are established protocols to isolate skin keratinocytes2,3,4 and fibroblasts5,6. These studies describe the isolation of either keratinocytes, fibroblasts, or both from the skin, but no protocol addresses how to establish cultures of primary SCs from human skin. Recent studies suggest that neuronal SCs modulate keratinocyte and fibroblast cellular processes and regulate normal skin physiological functions7. SCs are thus critical to skin homeostasis and contribute substantially to the regulate physiology that influences the behavior of neighboring skin cell types present8. Therefore, a protocol that allows for the isolation of each of these cell types is ideal for in vitro experiments involving cell-cell communication or cross-talk between cell types.
This protocol describes the establishment of individual cell cultures of primary cells from a single piece of skin. This protocol is particularly useful when the amount of tissue available is limited. Moreover, isolation of all three cell types from a single donor allows for robust comparisons between cell types or co-culture experiments while mitigating the influence of genetics during the desired experiment.
Protocol
Acquisition and use of de-identified human foreskin tissue for research purposes was reviewed and received the determination of "not human research" by the Penn State College of Medicine Institutional Review Board (IRB #17574).
NOTE: By following the protocol below, 2.4 x 106 keratinocytes, 4.4 x 106 fibroblasts and 1.1 x 106 SCs are obtained from a single foreskin. In general, these primary cells can be used for 3 passages, depending on the experimental conditions.
1. Isolation of primary cells and culture conditions
- Epidermis and dermis separation procedure
- Acquire neonatal foreskin following standard of care procedures by a physician under approved hospital regulatory protocols. The physician determines the timing of circumcision after birth and the amount of skin removed.
- Place the excised foreskin in a microcentrifuge tube containing 8-10 mL of ice-cold Dulbecco's Modified Eagle Medium (DMEM) basal medium and immediately transport the foreskin to the cell culture laboratory for cells isolation. Rinse the skin twice with 20-25 mL of 1x Dulbecco's phosphate-buffered saline (DPBS) containing 1x antibiotic/antimycotic.
NOTE: For maximum cell viability, the excised foreskin should be placed at 4 °C until processing, and cells should be harvested within 24 h. - After rinsing in DPBS/antibiotics, soak the foreskin in 25-30 mL of ice-cold DMEM basal medium for 10-15 min in a 10 cm tissue culture dish. Perform all processing steps in a tissue culture hood using sterile technique.
- Using a scalpel, carefully slice the foreskin open and expose the dermis and subcutaneous adipose tissue.
- Remove the subcutaneous adipose tissue using forceps, scalpel blade, and scissors, as needed. Discard the adipose tissue. Cut the cleaned foreskin into three to five smaller pieces.
- Transfer skin pieces to a sterile conical tube containing 16-20 mL of dispase-I/DMEM medium (4 mg/mL in DMEM basal medium).
- Mix the skin pieces in the conical tube by inversion, intermittently, during the first 30 min of incubation with dispase-I. Then place the conical tube at 4 °C for 16-18 h, being mindful of immersing the skin pieces in dispase-I/media.
NOTE: Dispase-I solution should be prepared just before addition using cold DMEM basal medium, filtered with a 0.22 µm strainer, and kept at 4 °C. - After overnight digestion with dispase-I, remove the skin pieces and place them on a dry, sterile, culture dish. Unroll each piece of skin so that the outer epidermal layer is touching the culture dish.
- Using two sets of forceps, carefully separate the epidermis from the dermis. Start on the edge of the tissue and peel the epidermis away from the dermis.
NOTE: If the epidermis layer is difficult to separate from the dermis, the dispase-I digestion was not sufficient. Troubleshooting options: 1) return skin pieces to dispase-I/DMEM solution and incubate for an additional 2-3 h at 4 °C; 2) place skin pieces in fresh dispase-I/DMEM and incubate at 4 °C for 2-3 h more. - Use the separated epidermis and dermis to isolate keratinocytes (epidermis) and fibroblasts and Schwann cells (dermis).
NOTE: Follow the steps below for specific cell types isolation and culture conditions.
- Keratinocyte isolation from the epidermis
- Add 5 mL of HEPES buffer saline (HBS) to a 10 cm culture dish and add the separated epidermis fragments. Incubate at room temperature (RT) for 10 min.
- After HBS incubation, collect the HBS solution in a 50 mL conical tube. Add 5 mL of trypsin neutralization buffer (TNB) (Table of Materials) in a 50 mL conical tube and set aside.
- Add 3 mL of trypsin solution to the epidermal fragments. Incubate at 37 °C for 10 min.
- Gently agitate the epidermal fragments using forceps until the trypsin solution becomes turbid.
- Add the trypsin solution/keratinocytes to the HBS and TNB containing tube in step 1.2.2.
- For any remaining epidermal fragments that have not dissolved in the initial trypsin digestion, add 3 mL of 0.25% trypsin/Ethylenediaminetetraacetic acid (EDTA). Incubate at 37 °C for 10 min and repeat steps 1.2.4 and 1.2.5.
- Add 2 mL of fetal bovine serum (FBS) to the trypsin/EDTA solution containing HBS and TNB solution tubes.
- Centrifuge the collection tube containing HBS/TNB/trypsin and keratinocytes at 870 x g for 5 min at 4 °C.
- Aspirate the supernatant and resuspend the cell pellet in 3 mL of keratinocyte complete growth medium (Table of Materials).
- Filter the cell suspension with a sterile 100 µM filter into a sterile 50 mL conical tube to remove debris.
- Quantify the cell number and cell viability.
NOTE: Here, a specialized cell sampling and staining device (Table of Materials) was used to quantify the cell number and viability following the manufacturer's instructions. - Seed culture plates as needed for experiments. The recommended seeding density is ~45,000 cells/cm2.
- Place the culture plates in an incubator at 37 °C in 5% CO2 for 2 days to allow keratinocytes to adhere to the dish.
- After 2 days, aspirate any non-adhered cells. Refresh the cell culture media every other day until keratinocytes reach desired confluence for passaging or downstream experiments (50%-80% confluence).
- Isolation of Schwann cells (SCs) and fibroblasts from the dermis
- Pre-coat T25 flasks with poly-L-lysine (PLL, 0.01 µg/µL) using 5 mL of 1x DPBS. Place the pre-coated flasks at 37 °C for 3 h. Wash the PLL coating matrix twice using 1x DPBS (5 mL).
NOTE: T25 flasks can be prepared ahead of time. - Rinse the isolated pieces of the dermis with 5 mL of DMEM basal medium.
- Mince the dermis into small pieces with scissors or a scalpel.
- Digest the minced dermis with 5 mL of collagenase (2 mg/mL in DMEM basal medium) at 37 °C for 2.5 h. Gently triturate the minced dermis with a pipette tip every 30 min until the dermis completely dissociates.
- Filter the cell suspension with a 70 µM cell strainer. It is necessary to apply mechanical force using a 1 mL syringe to fully filter the suspension.
- Dilute the filtered cell suspension with 5 mL of complete DMEM medium (DMEM basal medium + 5% FBS + 1x of antibiotic) to stop the enzymatic activity of collagenase.
- Centrifuge the cell suspension at 870 x g for 5 min at RT to pellet the cells.
- From this cell pellet, both fibroblasts and SCs will be obtained. Resuspend the cell pellet in 2 mL of DMEM complete medium to divide the cells (1 mL cell suspension/tube) and centrifuge at 870 x g for 5 min at RT.
- For SCs culture, follow steps 1.3.10-1.3.17 and for fibroblast culture, follow steps 1.3.18-1.3.22.
- From step 1.3.8, aspirate the supernatant. Resuspend the cell pellet in 5 mL of fresh DMEM complete medium.
- Quantify the cell number and viability.
NOTE: Here, a specialized cell sampling and staining device (Table of Materials) was used to quantify the cell number and viability following the manufacturer's instructions. - Seed the pre-coated T25 flasks with 5 mL of resuspended cells at a density 4.0 x 103 cells/mL. Incubate the flask at 37 °C in the presence of 5% CO2 overnight (~12-16 h incubation).
- After 16 h, confirm cell adhesion by microscopy (Table of Materials) at 10x magnification.
- Remove the non-adherent cells and wash the flask 3x with 5 mL of 1x DPBS.
- Add 5 mL of 10 µM cytosine arabinoside (antimitotic agent, kills fibroblasts) in DMEM complete medium. Incubate the flask at 37 °C in the presence of 5% CO2 for 24 h.
- Aspirate the cytosine arabinoside containing medium from the flask. Rinse the flask 3x with 5 mL of 1x DPBS.
- Refill the culture flask with 5 mL of SCs complete culture medium (Table of Materials) and incubate at 37 °C in the presence of 5% CO2 for 48 h. Refresh SC complete culture medium every other day until SCs reach 80% confluence.
- Aspirate the supernatant from the dissociated dermal layer (step 1.3.8) and resuspend the cell pellet with 5 mL of fibroblast complete medium (fibroblast basal medium + 5% FBS + 1x antibiotic).
- Quantify cell number and cell viability.
NOTE: Here, a specialized cell sampling and staining device (Table of Materials) was used to quantify the cell number and viability following the manufacturer's instructions. - Plate the fibroblasts at a cell density of 4.0 x 103 cells/mL in T25 culture flasks (not pre-coated) using 5 mL of fibroblast complete medium. Incubate at 37 °C in the presence of 5% CO2 overnight (~12-16 h incubation).
- After 16-24 h, confirm cell adhesion by microscopy (Table of Materials) at 10x magnification. Aspirate non-adherent cells from the flask and wash the flask twice with 5 mL of 1x DPBS.
- Add 5 mL of fresh fibroblast complete medium and incubate at 37 °C in the presence of 5% CO2 for 48 h. Refresh the culture medium every other day until fibroblasts reach the desired confluence for passaging or downstream assays (50%-80% confluence).
- Pre-coat T25 flasks with poly-L-lysine (PLL, 0.01 µg/µL) using 5 mL of 1x DPBS. Place the pre-coated flasks at 37 °C for 3 h. Wash the PLL coating matrix twice using 1x DPBS (5 mL).
2. Verification of epidermal keratinocytes, dermal SCs and fibroblasts protein marker by immunofluorescence
- Day 1- Coat chamber glass slides and detach cells
- Pre-coat 4-well chamber glass slides with collagen-I for keratinocytes, or PLL for SCs or, only 1x DPBS for fibroblasts (0.5 mL each well). Ensure to coat the entire surface. Incubate chamber slides at 37 °C for 2 h. During incubation, prepare the cells.
- After cultured cells reach 50%-80% confluence, wash the flasks with 1x DPBS 3x (1 mL for each well).
- Detach the adhered cells with 5 mL of 0.25% trypsin-EDTA (TE) solution by incubating at 37 °C in the presence of 5% CO2 for 2 min, until cells start to become round. Visualize cell detachment with microscopy (Table of Materials) at 10x magnification.
- After incubation, transfer TE solution containing cells to a conical tube with 5 mL fetal bovine serum (FBS).
- Add 5 mL of trypsin neutralization buffer (TNB) to the culture flask and then transfer the contents to the above-mentioned centrifuge tube (cells with TE and FBS). Visualize cell detachment with microscopy (Table of Materials) at 10x magnification.
NOTE: TNB or FBS or both can be used to neutralize trypsin activity. - Centrifuge the conical tube containing cells/TE/TNB at 870 x g for 5 min. Transfer the supernatant to another tube and resuspend the cell pellet in the corresponding complete culture medium. Measure the cell viability as mentioned in step 1.2.11.
- Rinse the pre-coated chamber glass slides (step 2.1.1) with 1x DPBS (1 mL for each well) 3x.
- Semi-dry the slides inside the tissue culture hood and seed keratinocytes, SCs, or fibroblasts at an appropriate density (30,000 cells/0.5 mL/chamber) in an appropriate complete cell culture medium.
- Incubate the chamber slides at 37 °C overnight to allow cells to adhere.
- Day 2- Blocking the chamber glass slide with Bovine Serum Albumin (BSA) and primary antibodies incubation
- Aspirate the cell media from the chamber slides and wash 3x with 1 mL of 1x DPBS.
- Fix the adhered cells with 4% paraformaldehyde in 1x DPBS (1 mL for each well) and incubate for 15 min at RT inside the tissue culture hood.
- Aspirate 4% paraformaldehyde from the chamber slides and wash the chamber slides 3x with 1x DPBS (1 mL for each well).
- Permeabilize the cell membranes with 1% TritonX-100 in 1x DPBS (1 mL for each well) and incubate for 15 min at RT.
- Aspirate the blocking solution from the chamber slides, wash 3x with 1x DPBS (1 mL for each well) for 30 s and then aspirate 1x DPBS from the well.
- Block the chamber slides with 5% BSA (0.5 mL for each well) and incubate for 45 min at RT.
- Aspirate the blocking solution from the chamber slides, wash 3x with 1x DPBS (1 mL for each well) for 30 s each and then aspirate 1x DPBS from the well.
- Dilute the primary antibodies with 5% BSA (0.2 mL for each well) (Primary antibodies dilutions: Keratinocytes: mouse cytokeratin 14 antibody (1:100) and, cytokeratin 10 antibody (1:100); Schwann cells: mouse S100 antibody (1:200), and rabbit nerve growth factor receptor (p75-NTR) antibody (1:500); Fibroblasts: rabbit vimentin antibody (1:200), and mouse alpha-smooth muscle actin (α-SMA) antibody (1:200)).
- Add the primary antibodies to the corresponding cells on the chamber glass slide and incubate for about 15-16 h (overnight) at 4 °C in a humidified chamber.
- Aspirate the primary antibodies from the chamber slides, wash 3 times with 1x DPBS (1 mL for each well) for 30 s each and then aspirate 1x DPBS from the well.
- Dilute the secondary antibodies (Goat anti-rabbit IgG Secondary Antibody-Alexa Fluor 488 (1:500) and Goat anti-Mouse IgG Secondary Antibody-Alexa Fluor 594 (1:500)) with 0.5% BSA. Add appropriate secondary antibody to each well (0.2 mL for each well). Incubate for 45 min at RT.
- Aspirate the secondary antibody from chamber slides, wash 3x with 1x DPBS (1 mL for each well) for 30 s each and then aspirate 1x DBPS from the well.
- Remove the gasket on the 4-well chamber slide using gasket remover. Try not to leave any adhesive on the slide as this interferes with coverslip application. Add a drop of mounting medium with 4',6-diamidino-2-phenylindole (DAPI). Alternatively, counter-stain the wells with DAPI and use a non-label-containing mounting medium.
- Carefully place the coverslip on the glass slide by avoiding air bubbles and seal the end of the coverslip with glue. Observe immunofluorescence staining patterns by microscopy (Table of Materials) at 10x and/or 20x magnification.
Representative Results
Normal neonatal foreskin was used for the isolation of primary epidermal keratinocytes and dermal SCs and fibroblasts. The isolated primary cells were cultured in respective cell culture media containing growth factors. After seeding of SCs and fibroblast in culture flasks, most of the cells adhered to the bottom of the flask within 2 h. In the case of keratinocytes, most keratinocytes adhered by 24 h. Isolated epidermal primary keratinocytes reached 85% confluence by day 7 and exhibited characteristic cell morphology (cobblestone-shape) (Figure 1A). The SCs reached 95% confluence by day 5 and appeared bi-polar or tri-polar in shape (Figure 1B). Fibroblast cultures reached 95% confluence by day 4 and most of the cells exhibited a spindle shape morphology (Figure 1C). Cell-type-specific protein expression was confirmed in these cultures by immunofluorescence staining. Keratinocytes were positive for K10 (differentiation marker) and K14 (proliferation marker) proteins (Figure 2A); similarly, SCs were positive for S100, and p75-NTR (Figure 2C). Fibroblasts positively expressed vimentin, but did not express the myofibroblast marker, alpha smooth muscle actin (α-SMA) (Figure 2E).
The merged images of keratin immunofluorescence and DAPI nuclear staining (Figure 2A) indicated that 97.8% of cells in the cultures were keratinocytes (Figure 2B). Double S100 and p75-NTR positive Schwann cells suggest purity of 95.2% SCs in culture following the protocol (Figure 2C,D). Similarly, 97.2% of cells in fibroblast cultures positively expressed vimentin (Figure 2E,F). Thus, characteristic protein expression is found in the respective cell types, and the protocol allows for the isolation of relatively pure populations of cells.
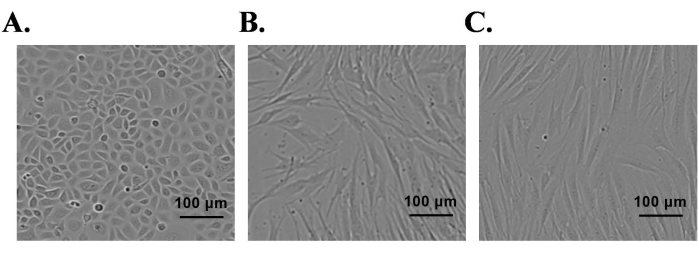
Figure 1: Isolation of human foreskin-derived primary keratinocytes, Schwann cells, and fibroblasts. (A) Keratinocytes, (B) Schwann cells, and (C) Fibroblasts under phase contrast microscope; scale bar = 100 µm. Please click here to view a larger version of this figure.
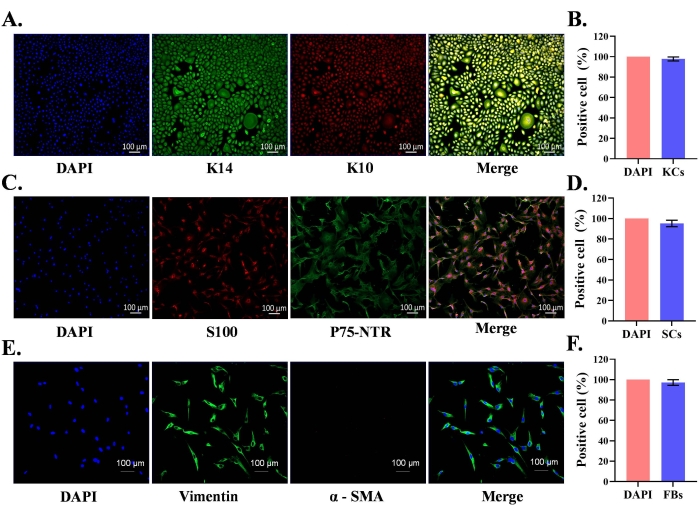
Figure 2: Characterization of human foreskin-derived primary keratinocytes, Schwann cells, and fibroblasts. (A) Keratinocytes were identified using keratin 14 (K14, proliferative keratinocytes) and K10 (keratin 10, keratinocyte differentiation marker); scale bar = 100 µm. (B) Statistical analysis of purity of cultured keratinocytes using immunofluorescences double stained cells. (C) Schwann cells were identified using S100 and p75-NTR (nerve growth factor receptor); scale bar = 100 µm. (D) Statistical analysis of purity of cultured SCs using immunofluorescences double stained cells. (E) Fibroblasts were identified using vimentin (fibroblast) and α-smooth muscle actin (fibroblast differentiation, myofibroblast); scale bar = 100 µm. (F) Statistical analysis of purity of cultured fibroblasts using immunofluorescences vimentin stained cells. Please click here to view a larger version of this figure.
Discussion
This protocol describes a method to isolate three distinct cell populations from a single piece of the foreskin, namely keratinocytes, fibroblasts, and Schwann cells. There are a few isolation protocols available to isolate keratinocytes and fibroblasts2,3,5,6, but none describe SC isolation. Apart from the key structural cells in skin, keratinocytes, and fibroblasts, skin is also highly innervated by sensory afferent structures and autonomic efferents. Sensory afferents play a significant role in the transmission of light touch, vibration, pain, itch, and hot and cold sensations. Autonomic efferents innervate sweat glands and arrector pili muscles9. Each nerve axon is ensheathed by a SC, the glial cells of the peripheral nervous system. Research and clinical interests in SCs have increased considerably as SCs support axonal regeneration10,11. Skin nerves are important regulators of skin physiology and disease pathology through communication signals (neuromodulators/neuromediators) to non-neural cells12,13,14.
Despite the wide distribution of nerve SCs in the skin, limited information exists regarding SCs specific role in skin physiology. How do SCs in the skin interact with other non-neuronal cells in the skin? Answering this question has been hindered by the lack of reliable in vitro cell culture models. Thus, with this protocol, the analysis of each individual cell and their interactions with cells from the same tissue can precisely reveal the characteristic features of Schwann cells' roles in skin homeostasis and pathophysiology.
One of the key advantages of this protocol is a simple, inexpensive experimental procedure to establish three primary cells (SCs, keratinocytes and fibroblast) from a single human foreskin. It also has the advantage of all three cell types being isolated from the same tissue, thus mitigating genetic differences in cell comparisons and optimizing cell isolations when tissue is in limited supply. This protocol takes 2 days to perform and simplifies the isolation procedure for three primary cells from skin tissue and culturing conditions. This protocol reliably yields high numbers of viable cells, and initial passages of these cells reached ~90% confluence by 4-7 days after initial plating. We have used keratinocytes for least 3 passages (~21 days) and fibroblast and Schwann cells can be used for 5-6 passages (~40 days in culture).
This protocol is not without technical challenges. Sufficient removal of the underlying adipose tissue and efficient epidermal/dermal separation before enzymatic digestion are key to obtaining relatively pure populations of cells from each skin layer. Dermal SCs are more difficult to isolate from the skin due to their relatively low prevalence in the skin compared to the more numerous fibroblasts. Any carryover of fibroblasts in SC cultures will rapidly outgrow SCs15. To enhance SC adherence, we opted to pre-coat tissue culture ware with PLL as this increases the adhesion of SCs16. To further mitigate fibroblast contamination, cytosine arabinoside (antimitotic agent) was added after cells had adhered for a short period of time to remove the rapidly dividing fibroblasts from SCs cultures. However, longer exposure to cytosine arabinoside can also be detrimental to SCs.
Adopting the above-described protocol makes it possible to isolate individual skin SCs, keratinocytes, and fibroblasts cultures for in vitro experiments. These distinct cell populations can be used to investigate SCs, keratinocytes, and fibroblasts individually or in combination during normal skin physiology, wound healing, or under conditions that mimic a disease setting.
Divulgations
The authors have nothing to disclose.
Acknowledgements
We would like to thank Dr. Fadia Kamal, and Dr. Reyad Elbarbary for allowing us to use lab instruments and technical support. This work was supported by grants from the NIH (K08 AR060164-01A) and DOD (W81XWH-16-1-0725) to J. C. E. in addition to institutional support from the Pennsylvania State University Hershey Medical Center.
Materials
| 0.22 µM sterile filters (Millex-GP Syringe Filter Unit,polyethersulfone) | MilliporeSigma | SLGPR33RS | |
| 70 µM cell strainers | CELLTREAT | 229483 | |
| 100 µM cell strainers | CELLTREAT | 229485 | |
| 1 mL disposable syringes | BD Luer-Lok | BD-309659 | |
| 5 mL disposable syringes (Syringe sterile, single use) | BD Luer-Lok | BD309646 | |
| 10 mL disposable syringes | BD Luer-Lok | BD305462 | |
| 1% TritonX-100 | Sigma | X100-1L | Prepared at the time of use |
| 4% paraformaldehyde solution | ThermoFisher Scientific | J19943.K2 | Ready to use and store at 4 °C |
| 5% BSA | Sigma | A7906-100G | Prepared at the time of use |
| 70% ethanol | Pharmco | 111000200 | |
| Antibiotic | ScienCell Research | 503 | |
| Chemometec Vial1-Cassette | Fisher Scientific | NC1420193 | |
| Collagenase | Gibco | 17018-029 | |
| Coverslip | Fisherbrand | 12544D 22*50-1.5 | |
| Dispase I | Sigma-Aldrich | D46693 | |
| DMEM basal medium | ScienCell Research | 9221 | |
| Dulbecco's phosphate-buffered saline free from Ca2+ and Mg2+ (DPBS) | Corning | 21-031-CV) | |
| Nunc 15 mL Conical Sterile Polypropylene Centrifuge Tubes | ThermoFisher Scientific | 339651 | |
| Nunc 50 mL Conical Sterile Polypropylene Centrifuge Tubes | ThermoFisher Scientific | 339653 | |
| 1.5 mL micro-centrifuge tubes | Fisherbrand | 02-681-5 | |
| Fetal bovine serum (FBS) | ThermoFisher Scientific | 10082147 | |
| Fibroblast complete medium | ScienCell Research | 2331 | |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Invitrogen | A11032 | Dilution (1:500) |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A11008 | Dilution (1:500) |
| Hanks' Buffered Saline Solution (HBSS buffer) | Lonza | CC-5022 | |
| Human foreskin | De-identified human foreskin tissue for research purposes (Institutional Review Board- IRB #17574). | ||
| KGM-GOLD keratinocyte medium (KGM gold and supplements) | Lonza | 00192151 and 00192152 | |
| Mouse alpha-smooth muscle actin antibody | ThermoFisher Scientific | 14-9760-82 | Dilution (1:200) |
| Mouse Cytokeratin14 antibody | Abcam | ab7800 | Dilution (1:100) |
| Mouse S100 antibody | ThermoFisher Scientific | MA5-12969 | Dilution (1:200) |
| Multi chambered (4 well glass slide) | Tab-Tek | 154526 | |
| NucleoCounter -Via1-Cassette | Chemometec | 941-0012 | |
| Poly-L-Lysisne (PLL) | ScienCell Research | 32503 | |
| ProLong Gold Anti-fade Mountant with DAPI | Invitrogen | P36935 | |
| Rabbit K10 antibody | Sigma-Aldrich | SAB4501656 | Dilution (1:100) |
| Rabbit p75-NTR antibody | Millipore | AB1554 | Dilution (1:500) |
| Rabbit vimentin | ProteinTech | 10366-1-AP | Dilution (1:200) |
| Schwann cell culture medium | ScienCell Research | 1701 | |
| Precision tweezers DUMONT straight with extra fine tips Dumostar, 5 | ROTH | LH75.1 | Sterilize with 70% alcohol before use |
| IRIS Scissors, sharp/sharp. Length 4–3/8"(111mm) | Codman | 54-6500 | Sterilize with 70% alcohol before use |
| Sterilized surgical – sharp blade (Duro Edge Economy Single Edge Blades) | Razor blade company | 94-0120 | Sterilize with 70% alcohol before use |
| T25 culture flask | Corning | 353109 | |
| Trypsin neutralization buffer (TNS) | Lonza | CC-5002 | |
| Trypsin/EDTA | Lonza | CC-5012 | |
| Inverted microscope | ZEISS | Axio Observer 7- Axiocam 506 mono – Apotome.2 microscope | For immunofluorescence of chamber slide containing stained cells |
| Inverted microscope | ZEISS | Primovert | For visulaizing/observing cell attachment or detachment |
References
- Hawksworth, G. M. Advantages and disadvantages of using human cells for pharmacological and toxicological studies. Human & Experimental Toxicology. 13 (8), 568-573 (1994).
- Green, H., Kehinde, O., Thomas, J. Growth of cultured human epidermal cells into multiple epithelia suitable for grafting. Proceedings of the National Academy of Sciences of the United States of America. 76 (11), 5665-5668 (1979).
- Fuchs, E., Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 19 (4), 1033-1042 (1980).
- Aasen, T., Izpisua Belmonte, J. C. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nature Protocols. 5 (2), 371-382 (2010).
- Seluanov, A., Vaidya, A., Gorbunova, V. Establishing primary adult fibroblast cultures from rodents. Journal of Visualized Experiments: JoVE. (44), e2033 (2010).
- Belviso, I., et al. Isolation of adult human dermal fibroblasts from abdominal skin and generation of induced pluripotent stem cells using a non-integrating method. Journal of Visualized Experiments: JoVE. (155), e60629 (2020).
- Silva, W. N., et al. Role of Schwann cells in cutaneous wound healing. Wound Repair and Regeneration. 26 (5), 392-397 (2018).
- Bray, E. R., Cheret, J., Yosipovitch, G., Paus, R. Schwann cells as underestimated, major players in human skin physiology and pathology. Experimental Dermatology. 29 (1), 93-101 (2020).
- Laverdet, B., et al. Skin innervation: important roles during normal and pathological cutaneous repair. Histology and Histopathology. 30 (8), 875-892 (2015).
- Jessen, K. R., Mirsky, R., Lloyd, A. C. Schwann cells: Development and role in nerve repair. Cold Spring Harbor Perspectives in Biology. 7 (7), 020487 (2015).
- Bentley, C. A., Lee, K. F. p75 is important for axon growth and Schwann cell migration during development. The Journal of Neuroscience. 20 (20), 7706-7715 (2000).
- Rutkowski, J. L., et al. Signals for proinflammatory cytokine secretion by human Schwann cells. Journal of Neuroimmunology. 101 (1), 47-60 (1999).
- Kumar, A., Brockes, J. P. Nerve dependence in tissue, organ, and appendage regeneration. Trends in Neurosciences. 35 (11), 691-699 (2012).
- Balakrishnan, A., et al. Insights into the role and potential of Schwann cells for peripheral nerve repair from studies of development and injury. Frontiers in Molecular Neuroscience. 13, 608442 (2020).
- Gresset, A., et al. Boundary caps give rise to neurogenic stem cells and terminal glia in the skin. Stem Cell Reports. 5 (2), 278-290 (2015).
- Stratton, J. A., et al. Purification and characterization of Schwann cells from adult human skin and nerve. eNeuro. 4 (3), (2017).
