Cladonema tentacles have been used as a model to study the cellular processes of morphogenesis and regeneration15,16,17. The tentacle structure is composed of an epithelial tube where stem-like cells, or i-cells, are located in the proximal region, called the tentacle bulb, and new branches are sequentially added to the rear of the distal region of the bulb along the adaxial side (Figure 3A)15. Previous reports have indicated that cell proliferation is active both in the tentacle bulb and at the new branching sites using either EdU or BrdU labeling16,17. However, due to the resolution of in situ hybridization, it is unclear whether stem-like cells are truly proliferative or not. To visualize both stem-like and proliferative cells simultaneously at the cellular level, we performed FISH for stem cell markers (Nanos1 or Piwi) and EdU labeling for S phase cells in the same samples.
At cellular resolution by FISH, the expression of Nanos1 was localized at the tentacle bulb and the new branching site (Figure 3B). Piwi was also expressed in the tentacle bulb and at the new branching site in a pattern similar to that of Nanos1 (Figure 3C). These results were consistent with the observations from whole-mount in situ hybridization in a previous report17, where the budding branch of a 7-day-old medusa was almost uniformly labeled by Nanos1 and Piwi. To visualize the beginning of the accumulation of stem-like cells, we monitored the new branching site of a 5-day-old medusa. The co-labeling of Nanos1 expression and EdU-positive cells revealed the spatial pattern of stem-like cells and proliferative cells in the tentacle (Figure 4A). Although the gross distributions of EdU+ and Nanos1+ cells were consistent with previous reports16,17, EdU+ cells were more widely distributed throughout the tentacle bulb, at least at the beginning of branching, while Nanos1+ cells accumulated more locally at the tentacle bulb and the new branching site (Figure 4A and Figure 4E). These observations suggest that distinct distributions of stem-like cells and proliferating cells are detected depending on the developmental timing and different stages.
A more detailed view of the bulb and the new branching site revealed that EdU signals merge with nuclear staining, whereas Nanos1 expression is constrained to the cytoplasm surrounding the nucleus, consistent with a previous report5 (Figure 4B,C). A fraction (19.79%) of the cells exhibited co-labeling of EdU and Nanos1 (EdU+ Nanos1+; Figure 4B,C, yellow arrowheads, and Figure 4D), suggesting that these cells are an actively-proliferating stem cell population. Intriguingly, 14.46% of the cells were found to be EdU+ Nanos1− in the middle of the bulb and at the new branching site, suggesting the presence of non-stem-like proliferative cells (Figure 4B,C, white arrows, and Figure 4D). In contrast, 26.32% of cells were observed to be EdU− Nanos1+ at the base of the bulb and at the new branching site, indicating the presence of a stem cell population that is either slow-cycling or quiescent, neither of which is detected by EdU pulse labeling (Figure 4B,C, yellow arrows, and Figure 4D).
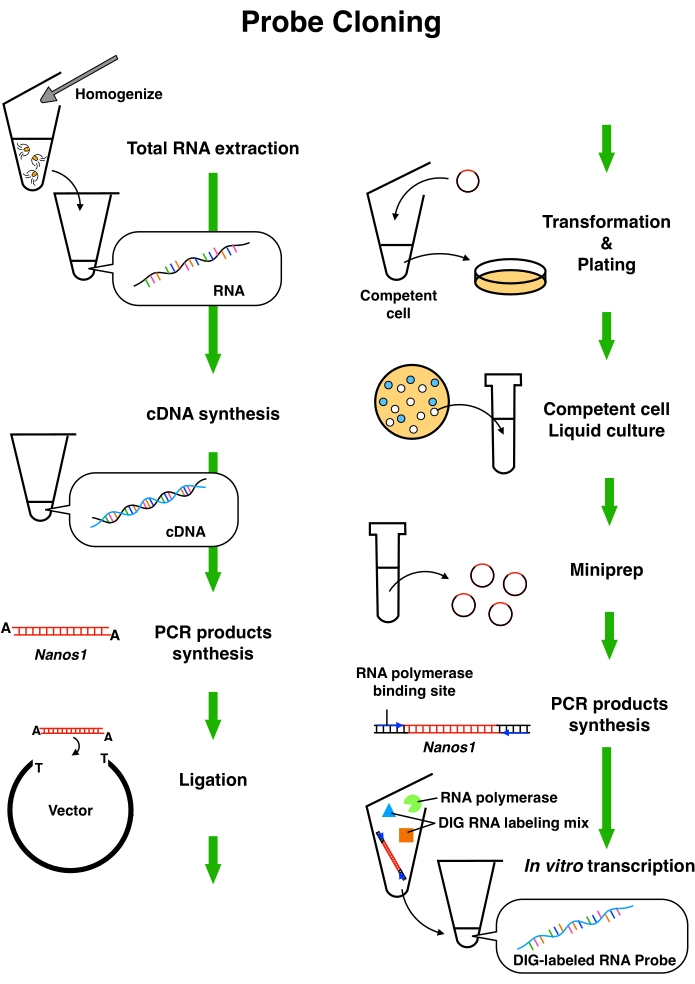
Figure 1: Scheme of the probe synthesis for in situ hybridization. Extraction of total RNA from medusae and cDNA synthesis from total RNA. Nanos1-specific PCR products were synthesized from the cDNA. The PCR products were ligated into the vectors, and amplified vectors were collected through competent cell culture. The PCR products with RNA polymerase binding sites were synthesized using the plasmids as templates. DIG-labeled RNA probes were synthesized by in vitro transcription. Abbreviations: DIG = digoxigenin. Please click here to view a larger version of this figure.
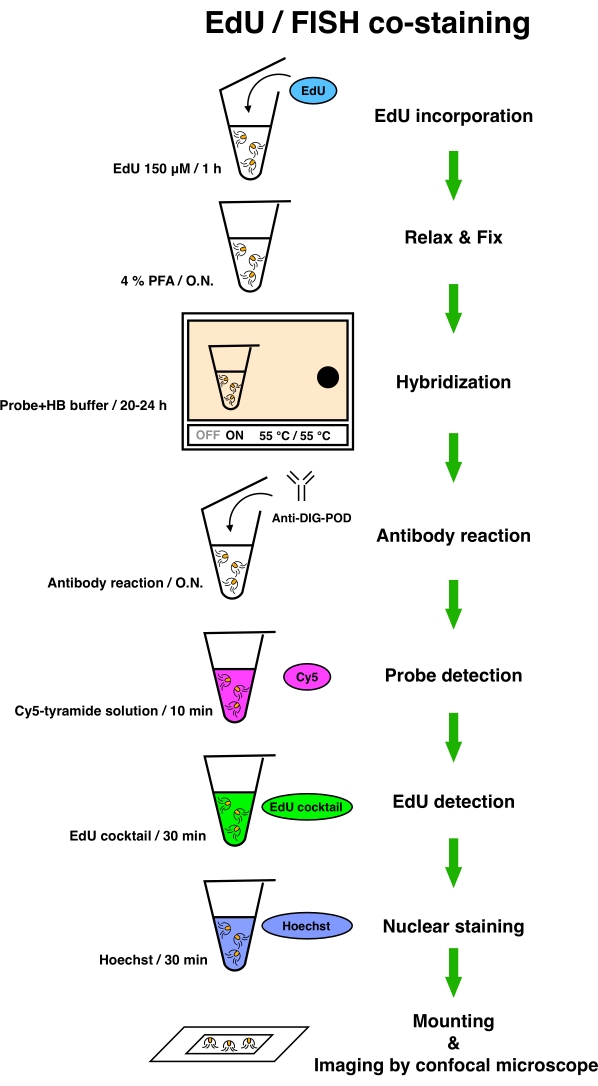
Figure 2: Scheme of EdU and fluorescent in situ hybridization co-staining. Medusae were incubated with 150 µM EdU for 1 h. Subsequently, the medusae were anesthetized (to relax the tissue) with 7% MgCl2 in H2O and fixed with 4% PFA O.N. at 4 °C. After fixation, the samples were hybridized with HB Buffer with a probe for 20-24 h at 55 °C. After the hybridization reaction, the samples were washed and incubated with anti-DIG-POD solution O.N. at 4 °C. The medusae were stained with Cy5-tyramide solution for 10 min, followed by the detection of EdU for 30 min and staining with Hoechst 33342 for 30 min. After completing all the staining processes, the medusae were mounted on the glass slide, and images were obtained with a confocal microscope. Abbreviations: EdU = 5-ethynyl-2'-deoxyuridine; FISH = fluorescent in situ hybridization; PFA = paraformaldehyde; O.N. = overnight; HB = hybridization. Please click here to view a larger version of this figure.
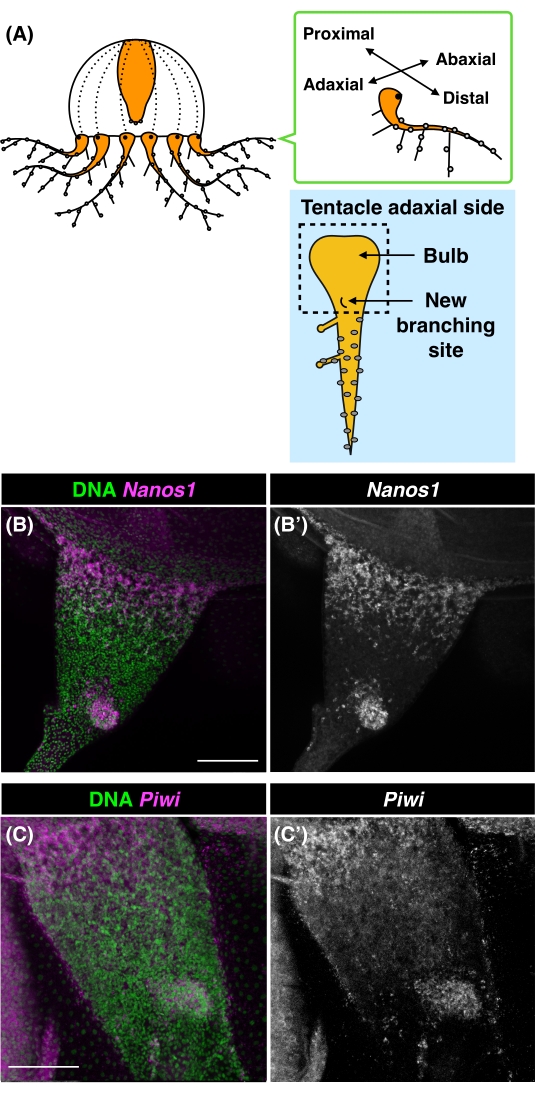
Figure 3: Nanos1 and Piwi expression patterns on the proximal adaxial side of the Cladonema medusa tentacle. (A) Schematic of a Cladonema medusa and tentacle. The adaxial side of the tentacle: the tentacle bulb (the most proximal region) with new branches sequentially formed on the new branching site. The inset (dashed square) indicates the area captured by the confocal image. (B) FISH images of Nanos1 gene expression from the proximal, adaxial side of the tentacle of a 7-day-old Cladonema medusa. (C) FISH images of Piwi gene expression from the proximal, adaxial side of the tentacle of a 7-day-old Cladonema medusa. DNA: green, Nanos1: magenta. B'–C' for Nanos1 FISH only images. Scale bars = 100 µm (B,C). Abbreviation: FISH = fluorescent in situ hybridization. Please click here to view a larger version of this figure.
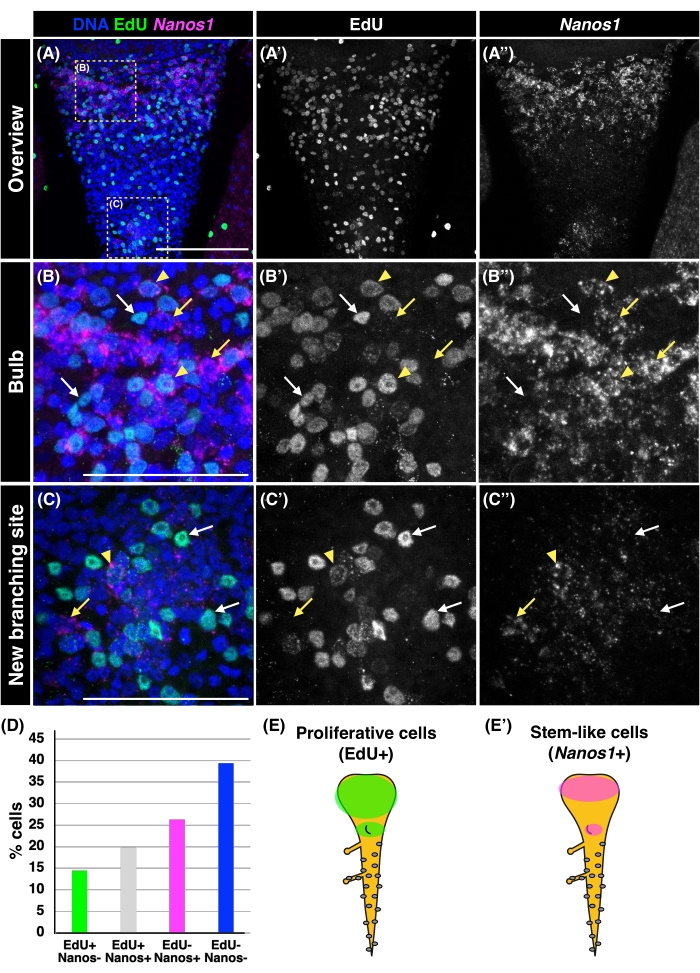
Figure 4: EdU and Nanos1 expression patterns on the proximal, adaxial side of Cladonema medusa tentacle. (A–C) Images of the proximal adaxial side of the Cladonema medusa tentacle co-labeled with Nanos1 expression and EdU; 5-day-old medusae were used. (A) An overview of the tentacle. Yellow dashed squares indicate the areas of B and C. (B) Magnification of the tentacle bulb. (C) Magnification of a new branching site. Yellow arrowheads indicate the cells that are positive for both EdU and Nanos1. Yellow arrows indicate the cells that are only positive for Nanos1. White arrows indicate cells only positive for EdU. A–C panels are merged images for DNA (blue), EdU (green), and Nanos1 (magenta). A'–C' are panels for EdU only images; A''–C'' are for Nanos1 FISH only images. Scale bars = 100 µm (A), 50 µm (B, C). (D) The quantification of EdU- and/or Nanos1-positive cells in the basal side of the tentacle (quantification area = 30.10 µm2 square, n = 6, a total of 249 cells). EdU+ Nanos1− cells, 14.46%; EdU+ Nanos1+ cells, 19.79%; EdU− Nanos1+ cells, 26.32%; EdU− Nanos1− cells, 39.44%. (E) Schematic of a Cladonema medusa tentacle from the adaxial side.The overall distribution of EdU+ cells and Nanos1+ cells are shown in E and E', respectively. Please click here to view a larger version of this figure.
Table 1: Composition of different PCR reactions and buffers in this protocol. To calculate the volume of the PCR product (X µL) in the ligation reaction, see the note after protocol step 1.4. Please click here to download this Table.
