1. Making polyvinyl alcohol (PVA) solution
- Measure water and PVA polymer pellets (see Table of Materials) to create a 10 mL solution at a 20% PVA to water ratio by weight.
- Heat the water in the glassware over a hot plate set to 100 °C.
- Place the PVA polymer pellets into the heated water. Insert a magnetic stir bar.
- Reduce the heat to 80 °C and stir until the PVA fully dissolves.
- Cover the top of the glassware to prevent contamination.
- Once fully dissolved, place the 20% PVA solution into an appropriate storage container for storage at room temperature.
2. Preparing PVA-coated silicon (Si) wafers
- Cut a silicon (Si) wafer (see Table of Materials) into ~10 x 10 mm2 squares.
- Clean the Si substrate using isopropyl alcohol and let it dry.
- Place the clean Si wafer on the chuck of the spin coater (see Table of Materials).
- Drop cast approximately 10 µL of PVA solution onto the center of the Si substrate. Try to avoid bubble formation.
- Coat the Si substrate with a uniform PVA film by spin coating for 30 s at 1,500 rotations per minute (rpm).
NOTE: The specified volume of liquid and spin coating parameters create a uniform layer of PVA across the surface of the substrate with sufficient thickness to prevent rapid drying between the spin coating and placing the PS beads onto the PVA surface in the next step.
- Remove the sample from the spin coater and place it into a clean sample container to prevent contamination before transferring the PS beads.
3. Placing the PS beads onto the PVA-coated surface
- Clean a Si substrate using isopropyl alcohol and let it dry.
- Using a pipette, place 1 µL of PS beads suspended in water onto the center of the substrate.
- Let the water evaporate by placing the sample in a storage compartment containing bentonite clay desiccant.
NOTE: This step preserves the sample by reducing exposure to humidity. - Place the PVA-coated substrate (step 2.4) and the substrate with the dried PS beads (step 3.3) under an optical microscope. Depending on their size, a single bead will be visible using simple binoculars or will require higher optical magnification.
- Gently loosen the beads using ultrafine tweezers (see Table of Materials). Use a fine hair paintbrush to collect a few loose beads and lightly tap the hairs of the paintbrush over the freshly PVA-coated wafer. Multiple sweeps should allow the beads to accumulate within the hairs of the brush. Tap the top of the paintbrush hairs to disturb the PS bead powder to release beads onto the tacky PVA surface.
NOTE: It is important that the paintbrush is of high quality and clean to avoid releasing fibers and contaminants onto the PVA film's surface. Moving quickly during this step is essential so that the PVA does not completely dry. - Repeat this step until it is confirmed by optical microscopy inspection that individual PS beads adhere to the PVA surface.
- Store the sample in a clean container. Allow the sample to dry fully.
NOTE: The sample should be allowed to cool and dry completely before further analysis attempts are performed. AFM height measurements or surface profiler measurements can assess the thickness of successive PVA films.
4. Loading the sample for AFM characterization
NOTE: The protocol described is based on standard operating procedures of a nanoIR2 (see Table of Materials) platform, but should be adapted according to the AFM model used for the measurement.
- Mount the PVA and PS bead sample onto the AFM stage using a metallic AFM disk and adhesive tabs, so that the sample is firmly attached to the sample holder.
- Mount a nanoIR probe (e.g., FORTGG) onto the AFM probe holder.
NOTE: The AFM cantilever is 225 µm long, 27 µm wide, and 2.7 µm thick, with a tip radius of less than 10 nm. The cantilever is coated with 45 nm thick gold film on both sides to limit its response to the top-side IR illumination of the sample (see Table of Materials). For nanoIR spectroscopy measurements, preferably use a cantilever that has been stored in a polydimethylsiloxane-free environment prior to use. - Align the read-out laser at the free end of the cantilever beam by turning the laser alignment knobs (x and y adjustments of the laser position and vertical adjustment of the detector position).
- Maximize the SUM signal of the detector.
- Adjust the position of the detector by turning the deflection knob so that the laser is aligned with the center of the position-sensitive detector of the AFM read-out system, corresponding to a vertical deflection signal of ~0 V.
- Click on the Load icon in the AFM "Probe" control panel.
- Follow the prompts within the wizard screen. Use the focus arrows to determine the focal plane of the nanoIR cantilever. Use the XY-displacement controls to position the cantilever in the center of the screen (aligned with the white cross).
- Next, click on the focus arrows to find the focal plane of the surface of the sample.
- Use the optical view of the system and the XY-displacement controls to position the cantilever tip above the bead of interest and click on Next.
- On the engage screen, set the "standoff" to 50 µm and click on Approach Only.
- Initiate the Engage procedure to approach the tip for imaging.
5. Creating topographical and nanoIR images of the multipolymer sample
- Acquire topography images in standard "Contact Mode". Once the position of the cantilever with respect to the PS bead is set, initiate the approach by clicking on the engage icon in the AFM "Probe" control panel. An engage setpoint of a deflection differential of 0.2 V is used for the entire study here, corresponding to a force of ~100 nN.
In the AFM "Scan" control panel, set the scan rate to 0.8 Hz, scan size (height and width), and the number of points per line and number of lines per image to use for imaging (512 x 512 was used here). Click on Scan to acquire the topography image.
NOTE: Calibration of the cantilever18 is done by determining the deflection sensitivity (in nm/V) from the slope of the deflection-distance curve obtained with the cantilever interacting with a sapphire substrate (Supplementary Figure 1A). The cantilever stiffness is determined from thermal tuning19 (Supplementary Figure 1B). The resonance of the cantilever is fitted using a Lorentzian function. The cantilever stiffness (in N/m) is determined using the equipartition theorem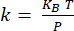 , where KB is the Boltzmann constant, T is the temperature (T = 295K), and P is the area of the power spectrum of the thermal fluctuations of the cantilever determined by integrating the Lorentzian fit of the thermal tuning data20.
, where KB is the Boltzmann constant, T is the temperature (T = 295K), and P is the area of the power spectrum of the thermal fluctuations of the cantilever determined by integrating the Lorentzian fit of the thermal tuning data20. - For nanoIR measurements, position the AFM tip on the feature of interest identified from the topography image.
- Select the tuning fork icon in the nanoIR control panel to determine the contact resonance frequencies of the cantilever. Set an illumination wavenumber that will excite photothermal expansion in the material. Set a range of laser pulse frequency to sweep and set the duty cycle of the nanoIR laser. Select Acquire within the "Laser Pulse Tune Window".
- Select the second contact resonance of the tip-sample system for nanoIR measurements by positioning the marker bar (green vertical line) at the peak of the second contact resonance.
NOTE: The selection of the contact resonance mode can vary depending on the type of cantilever and sample. - Click on Optimize to align the center of the IR laser focal region with the position of the cantilever tip. Alignment is done at a selected IR illumination wavenumber, corresponding to an absorption band of the material probed. The cantilever is positioned at the center of the laser footprint (Supplementary Figure 2).
NOTE: Alignment can vary for different wavenumbers depending on the laser model. - Acquire a background of the IR laser illumination. This consists of measuring the output of the IR quantum cascade laser (QCL) in the wavelength range of emission at the pulse frequency selected (Supplementary Figure 3). This is important for background correction of the nanoIR spectra.
- Acquire the nanoIR spectrum by selecting the wavenumber range (here, Start and Stop are set to 1,530 cm-1 and 1,800 cm-1, respectively), the step size (2 cm-1), and the number of averages used for the measurement. Perform background correction of the spectra displayed by dividing the photothermal amplitude measured by the attenuated background, which consists of multiplying the background collected in step 5.2.4 by the percentage of power selected for the measurement.
- For nanoIR imaging, select the region of interest for imaging.
- Enable phase-locked loop (PLL) auto-tune in the "Laser Pulse Tune Window" (accessed by the Tuning fork icon).
- Adjust the minimum and maximum frequency to create a sweep range centered at the second resonance mode in the general control panel.
- Zero the phase by clicking on zero in the PLL control panel and then click on OK in the "Laser Pulse Tune window".
- Select IR Imaging Enabled by putting a checkmark in the box within the nanoIR control panel.
- In the "Imaging View" control panel, choose Hauteur (Imaging View 1), Amplitude 2 (Imaging View 2), and Phase 2 (Imaging View 3) to acquire the topographical and chemical images of the sample. Set the acquisition direction to Trace (or Retrace). A line fit Line is often required to observe the topography image of the sample being acquired. Capture fit should be set to None.
NOTE: Scanning preferences, such as scan direction captured or color palette used, can be adjusted as needed. - In the AFM "Scan" control panel, select the Scan icon.
- To save the image, select the Now or End of frame icon in the "Capture" control panel.
- To export the data, right-click on the image or spectrum file names within the data lists. Select Export and choose the format of the file to export. Save the file in the desired computer folder location.
PS ((C8H8)n) beads were deposited on a clean Si substrate (Figure 1A) and on PVA ((CH2CHOH)n) (Figure 1B,C). Due to the poor adhesion of the bead on Si, nanoIR imaging in contact mode could not be acquired for this sample. Instead, using the optical view of the sample on nanoIR, the gold-coated AFM probe was engaged on top of the PS bead in contact mode, with an estimated force of about 100 nN (Figure 2A). The pulsed IR laser was set to excite the sample at 1,730 cm-1, since both PS and PVA are expected to absorb at this wavenumber. The pulse frequency of the laser was swept to determine the contact resonance of the cantilever for the nanoIR spectrum measurement. When pulsed at the contact resonance frequency, the cantilever response was monitored to determine the amplitude of the oscillation (Figure 2B). Next, the nanoIR spectra were constructed by monitoring the amplitude of the contact resonance as a function of the illumination wavenumber, from 1,530 to 1,800 cm-1 in steps of 2 cm-1 (Figure 2C). In this range (Figure 3A, inset), the spectrum revealed the presence of two IR bands centered at 1,600 cm-1 and 1,730 cm-1, corresponding to the predominant stretch mode of the phenyl moiety and a subset of the ring stretching in PS, respectively21. Comparing the nanoIR spectrum with the far-field Fourier transform infrared (FTIR) spectrum of PS confirmed the presence of the aromatic mode at 1,600 cm-1 (Figure 3A). However, it was noted that the relative amplitude I1600/I1730 was significantly different in the FTIR and nanoIR spectra, with respective values of 2.9 and 0.9. This was attributed to the mechanism of detection of nanoIR spectroscopy, which monitors the photothermal expansion of the polymer instead of its absorbance, as in FTIR spectroscopy. In the case of PS, this corresponded to a higher photothermal expansion when exciting the aromatic sub-bands at 1,730 cm-1. The nanoIR spectrum of PVA exhibited better agreement with the FTIR spectrum with a predominant absorption band centered at ~1,730 cm-1 (Figure 3B). While this band was not expected in pure PVA, which does not contain a C=O group, prior work suggests that the presence of the band can be attributed to carbonyl functional groups due to residual acetate used in the preparation of PVA (rate as ~80%-90% hydrolyzed)22. For the purpose of this study, the presence of a band at 1,730 cm-1 was suitable to assess the effect of simultaneous absorption of PS and PVA.
The nanoIR spectra were used to select the illumination wavenumbers for chemical imaging of the PS bead deposited at the surface of PVA (Figure 4A–C) and of the PS bead coated with PVA (Figure 4D–F). The PLL contact resonance frequency tracking capability of the system was used to ensure that the amplitude measurement at each pixel corresponded to the maximum amplitude of the contact resonance peak15 (Figure 2C). The contact resonance of the cantilever was measured when the cantilever tip was in contact with PVA and PS to determine a suitable range of the PLL frequency tracking.
NanoIR images were first acquired at 1,600 cm-1, which corresponds to the case where PS is the predominant absorber of the system (Figure 4Ai; Di)). Although a 5 µm PS bead was imaged in both cases, the amplitude of the photothermal expansion recorded at this wavelength was different when the tip was directly in contact with the PS bead and when the tip was in contact with the thin PVA coating on top of the PS bead. The increase in photothermal expansion detected above the PS bead was significantly smaller when the bead was covered with the layer of PVA, estimated to be ~1.8 µm thick using a surface profiler (Supplementary Figure 4). At the laser power used (1.47%, which corresponds to ~3.4 mW at 1,600 cm-1 and ~3.8 mW at 1,730 cm-1) (Supplementary Table 1), a region ~13 µm in diameter exhibited a slight increase in amplitude ~2 nm above the signal recorded on the pure non-absorbing PVA layer away from the PS bead. The spatial footprint of the amplitude increase was much broader than when the PS bead was on top of the PVA film (Figure 4A), where the photothermal expansion signal was asymmetrical but remained contained within a region ~6 µm wide in the fast scan direction and ~8 µm long in the slow scan direction of the image. The amplitude readings in this region reached up to 12.1 nm above the photothermal expansion of the pure PVA layer. When illuminating the sample at 1,730 cm-1, PS and PVA both exhibited a higher amplitude of photothermal expansion than at 1,600 cm-1. In the case of the exposed PS bead (Figure 4Ci), the photothermal expansion was highest on top of the bead, reaching values up to 26.5 nm. The high photothermal response extended several micrometers beyond the footprint observed in Figure 4Ai. The PVA also expanded due to the excitation, but at a lower amplitude, namely ~7.6 nm recorded away from the PS bead. It was noted that the response was consistently strongest on the left side of the PS bead. For the embedded bead (Figure 4F), the signal was more symmetrical, but the amplitude of the photothermal expansion above the PS bead was only ~2.3 nm higher than that of the PVA. For this, a consistent photothermal amplitude of ~13.6 nm was recorded within the 10-13 µm footprint of the region affected by PS heating, determined from mapping the surface with the AFM probe. In the case of measurements carried out at 1,620 cm-1, no signal was detected, which is consistent with the absence of a notable IR absorption band for PS and PVA in this region of spectrum.
Next, the nanoIR spectra collected on top of a PS bead covered was compared with PVA (Figure 5A). The PS signal at 1,600 cm-1 was significantly lower than in the case of the uncovered bead. Despite the low contribution of the band, further analysis of the signals confirmed that increasing the laser power led to a higher ratio I1600/I1730 (Figure 5B,C). The results suggested that a higher laser power corresponds to larger penetration depth, as depicted in Figure 5B. In turn, this affects the nanoIR image collected under a different laser power. At a higher power of ~20 mW, the photothermal amplitude of the material consistently exhibited a lower amplitude than at lower laser powers. Further, the noise level in the spectra increased, suggesting some instabilities in the material, likely due to the increase in temperature in the polymer.

Figure 1: Deposition of PS beads. Scanning electron microscopy (SEM) image of the 5 µm PS bead (A) on top of a pristine silicon substrate, (B) on top of a PVA film, and (C) covered with PVA. Scale bar = 1 µm. Please click here to view a larger version of this figure.
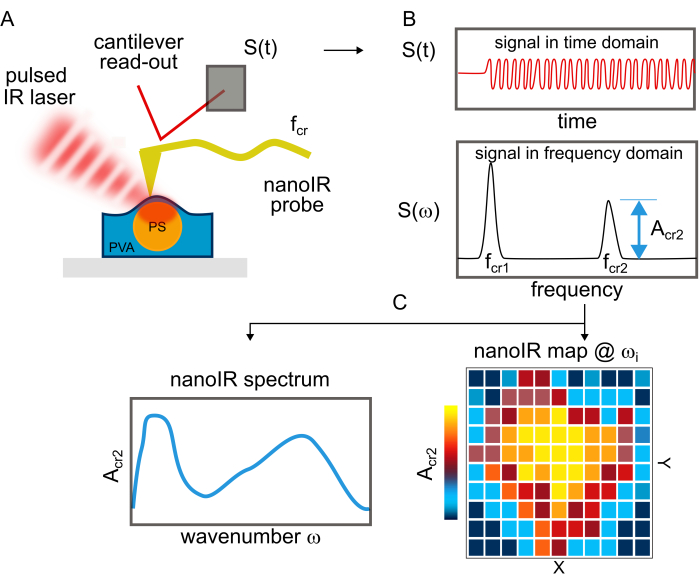
Figure 2: Schematics illustrating the principle of nanoIR imaging and spectroscopy. (A) The IR pulsed laser is aligned and focused onto the point of contact of the metallic AFM cantilever tip with the sample. The cantilever measures the photothermal response of the material resulting from IR absorption. The gold layer on both sides is used to reduce the photothermal and acoustic contribution of the cantilever during the measurement, which would interfere with the nanoIR measurement of the sample. (B) Signal of the position-sensitive detector monitoring the deflection of the cantilever in the time domain [S(t)] and frequency domain [S(ω)]. The contact resonance modes of the cantilever are identified by lock-in amplifier measurements. The second contact resonance is used for PLL tracking during imaging. The amplitude of the contact resonance mode Acr2 is measured. (C) NanoIR spectrum obtained by monitoring the change in amplitude Acr2 as a function of illumination wavenumber. The NanoIR image was obtained by monitoring the change in amplitude Acr2 as a function of the position of the tip on the sample. Please click here to view a larger version of this figure.
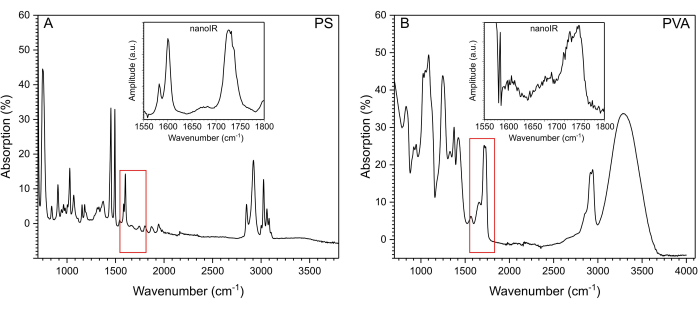
Figure 3: IR spectra of PS and PVA. (A) FTIR spectrum of PS. The red box indicates the range of the spectrum studied with nanoIR spectroscopy. The corresponding nanoIR PS spectrum is provided in the inset. (B) FTIR spectrum of PVA. The red box indicates the range of the spectrum studied with nanoIR spectroscopy. The corresponding nanoIR PVA spectrum is provided in the inset. Please click here to view a larger version of this figure.
Figure 4: NanoIR imaging. The PS bead (A–C) deposited on the surface of PVA and (D–F) embedded in PVA as depicted in the insets. NanoIR maps acquired at (Ai,Di) 1,600 cm-1, (Bi,Ei) 1,620 cm-1, and (Ci,Fi) 1,730 cm-1 are overlayed on the 3D topography map. Corresponding topography and nanoIR profiles extracted along the white dashed line are presented in the respective graphs (ii). Scale bar = 5 µm. Please click here to view a larger version of this figure.
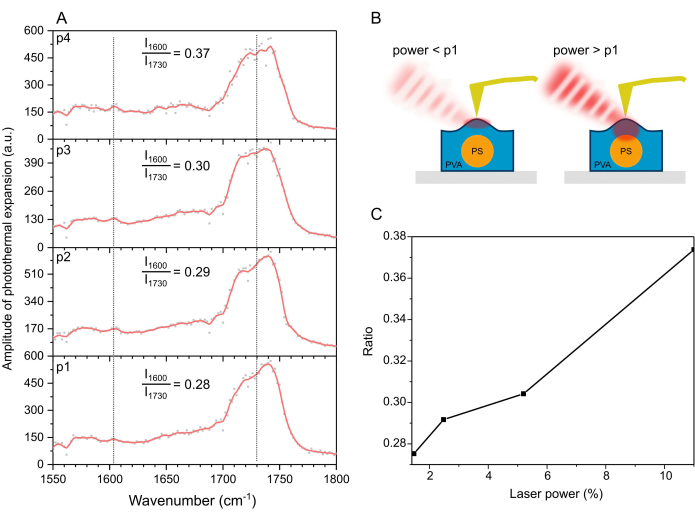
Figure 5: NanoIR spectrum collected by the AFM tip in contact with the PS bead coated with PVA. (A) NanoIR spectra collected at p1 = 1.47%, p2 = 2.48%, p3 = 5.20%, and p4 = 11% of the laser power. Laser power varies as a function of wavelength, which is corrected for by attenuated background correction for each case. (B) Schematics illustrating the effect of laser power on the strength of the signals collected. (C) Ratio I1600/I1730 as a function of laser power. Please click here to view a larger version of this figure.
Supplementary Figure 1: Calibration of the nanoIR cantilever. (A) Deflection-distance curve obtained on a sapphire substrate. The slope of the retract curve provides the deflection sensitivity of the cantilever used to calculate the spring constant. (B) Cantilever resonance frequency determined by thermal tuning used to calculate the spring constant. Please click here to download this File.
Supplementary Figure 2: IR laser footprint. The oscillation amplitude of the cantilever in contact with the sample surface is recorded as a function of the position of the IR laser. The IR laser alignment is varied using a moving mirror. The position of the mirror is set to align the center of the focal volume (red region) with the position of the cantilever (center of the white cross). Please click here to download this File.
Supplementary Figure 3: Laser output power (in mW) as a function of wavenumber. The output power of the QCL laser is determined using a standard IR detector from 1,530 cm-1 to 1,800 cm-1 in steps of 2 cm-1 at the pulse frequency corresponding to the setting used for the nanoIR measurements. Please click here to download this File.
Supplementary Figure 4: PVA thickness measurements obtained by laser confocal surface profilometry. Please click here to download this File.
Supplementary Table 1: The laser power in milliwatts for each wavenumber. Please click here to download this File.
