Lipiden zijn belangrijke bestanddelen van membranen, die grenzen bieden aan cellen en intracellulaire compartimentering mogelijk maken 1,2,3. Lipiden zijn amfifiel, met een polaire kopgroep en twee hydrofobe vetzuurstaarten; Deze assembleren zichzelf tot een dubbellaag om het contact van de hydrofobe ketens met water te minimaliseren 3,4. Verschillende combinaties van hydrofiele kopgroepen en hydrofobe staarten resulteren in verschillende klassen lipiden in biologische membranen, zoals glycerofosfolipiden, sfingolipiden en sterolen (Figuur 1)1,5,6. Glycerofosfolipiden zijn primaire bouwstenen van eukaryote celmembranen die zijn samengesteld uit glycerofosfaat, vetzuren met lange ketens en hoofdgroepen met een laag moleculairgewicht7. De lipidennomenclatuur is gebaseerd op verschillen in hoofdgroepen; voorbeelden zijn fosfatidyl-choline (PC), fosfatidyl-ethanolamine (PE), fosfatidyl-serine (PS), fosfatidyl-glycerol (PG), fosfatidyl-inositol (PI) of het niet-gemodificeerde fosfatidinezuur (PA)5,6. Wat hydrofobe staarten betreft, variëren de lengte en mate van verzadiging, samen met de ruggengraatstructuur. De mogelijke combinaties zijn talrijk, wat resulteert in duizenden lipidensoorten in zoogdiercellen6. Veranderingen in de samenstelling van membraanlipiden leiden tot verschillende mechanische en structurele membraaneigenschappen die van invloed zijn op de activiteit van zowel integrale membraaneiwitten als perifere eiwitten 2,6.
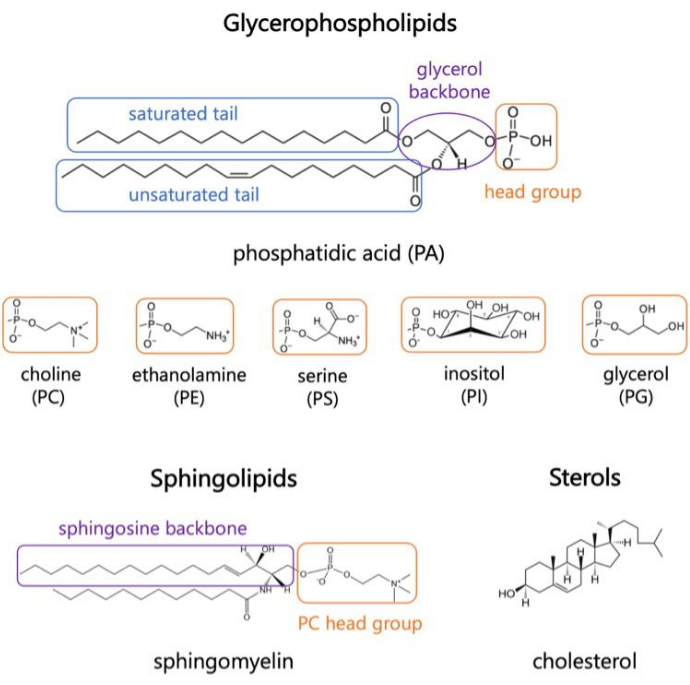
Figuur 1. Representatieve lipidestructuren. Vetzuurstaarten worden weergegeven in blauwe vakken, gemeenschappelijke lipidekopgroepen in oranje en monsterruggengraat in paars. Klik hier om een grotere versie van deze figuur te bekijken.
Lipiden spelen een actieve rol in cellulaire processen, eiwitactivering in signaalcascades en homeostase van gezonde cellen 8,9. Veranderde lipidendynamiek is het gevolg van infectie of kan markers zijn van pathogenese van ziekte 10,11,12,13,14,15. Als barrières voor de cel is de studie van membraanlipiden en hun rol in de permeatie van kleine moleculen van belang voor medicijnafgiftesystemen en membraanverstoringsmechanismen16,17. Chemische diversiteit en verschillende verhoudingen van lipidesoorten in organellen, weefsels en organismen geven aanleiding tot complexe membraandynamieken2. Het is daarom belangrijk om deze kenmerken te behouden in modelleringsstudies van lipidedubbellagen, vooral wanneer het doel van een studie is om interacties van andere biomoleculen met het membraan te onderzoeken. De lipidensoorten waarmee in een model rekening moet worden gehouden, hangt af van het organisme en het cellulaire compartiment dat van belang is. PG-lipiden zijn bijvoorbeeld belangrijk voor elektronenoverdracht in fotosynthetische bateria18, terwijl gefosforyleerde inositollipiden (PIP’s) belangrijke spelers zijn in de dynamiek van plasmamembraan (PM) en signaalcascades in zoogdiercellen 19,20. In de cel bevatten de PM-, endoplasmatisch reticulum (ER), Golgi en mitochondriale membranen unieke lipide-abundanties die hun functie beïnvloeden. Het ER is bijvoorbeeld de hub voor lipidenbiogenese en transporteert cholesterol naar de PM en Golgi; het bevat een hoge lipidendiversiteit met een overvloed aan PC en PE, maar een laag sterolgehalte, wat de vloeibaarheid van het membraan bevordert21,22,23,24. Daarentegen bevat de PM honderden en zelfs duizenden lipidensoorten, afhankelijk van het organisme25, het bevat een hoog gehalte aan sfingolipiden en cholesterol die het een karakteristieke stijfheid geven in vergelijking met andere membranen in de cel24. Bladasymmetrie moet worden overwogen voor membranen zoals de PM, die een buitenste bijsluiter heeft die rijk is aan sfingomyeline, PC en cholesterol, en een binnenblad dat rijk is aan PE, PI en PS die belangrijk zijn voor het signaleren van cascades24. Ten slotte leidt lipidendiversiteit ook tot de vorming van microdomeinen die verschillen in verpakking en interne volgorde, bekend als lipide vlotten24,26; Deze vertonen laterale asymmetrie, er wordt verondersteld dat ze een belangrijke rol spelen in cellulaire signalering26 en zijn moeilijk te bestuderen vanwege hun voorbijgaande aard.
Experimentele technieken zoals fluoroscopie, spectroscopie en modelmembraansystemen zoals gigantische unilamellaire blaasjes (GUV’s) zijn gebruikt om interacties van biomoleculen met membranen te onderzoeken. De complexe en dynamische aard van de betrokken componenten is echter moeilijk vast te leggen met experimentele methoden alleen. Er zijn bijvoorbeeld beperkingen aan de beeldvorming van transmembraandomeinen van eiwitten, de complexiteit van membranen die in dergelijke onderzoeken worden gebruikt, en de identificatie van tussenliggende of voorbijgaande toestanden tijdens het proces van belang27,28,29. Sinds de komst van de moleculaire simulatie van lipide monolagen en dubbellagen in de jaren 198029, kunnen lipide-eiwitsystemen en hun interacties nu op moleculair niveau worden gekwantificeerd. Moleculaire dynamica (MD)-simulatie is een veelgebruikte computertechniek die de beweging van deeltjes voorspelt op basis van hun intermoleculaire krachten. Een additieve interactiepotentiaal beschrijft de gebonden en niet-gebonden interacties tussen deeltjes van het systeem30. De set parameters die wordt gebruikt om deze interacties te modelleren, wordt het simulatiekrachtveld (FF) genoemd. Deze parameters worden verkregen uit ab initio-berekeningen, semi-empirische en kwantummechanische berekeningen, en geoptimaliseerd voor gereproduceerde gegevens van röntgen- en elektronendiffractie-experimenten, NMR, infrarood, Raman- en neutronenspectroscopie, naast andere methoden31.
MD-simulaties kunnen worden gebruikt om systemen op verschillende resolutieniveaus te bestuderen32,33,34. Systemen die gericht zijn op het karakteriseren van specifieke biomoleculaire interacties, waterstofbruggen en andere details met hoge resolutie worden bestudeerd met all-atom (AA) simulaties. Grofkorrelige (CG) simulaties daarentegen klonteren atomen in grotere functionele groepen om de rekenkosten te verlagen en de dynamiek op grotere schaal te onderzoeken33. Tussen deze twee bevinden zich united-atom (UA) simulaties, waarbij waterstofatomen worden gecombineerd met hun respectievelijke zware atomen om de berekening te versnellen33,35. MD-simulaties zijn een krachtig hulpmiddel voor het verkennen van de dynamiek van lipidemembranen en hun interacties met andere moleculen en kunnen dienen om mechanismen op moleculair niveau te bieden voor processen die van belang zijn op het membraangrensvlak. Bovendien kunnen MD-simulaties dienen om experimentele doelen te verfijnen en macromoleculaire eigenschappen van een bepaald systeem te voorspellen op basis van microscopische interacties.
In het kort, gegeven een reeks initiële coördinaten, snelheden en een reeks omstandigheden zoals constante temperatuur en druk, worden posities en snelheden van elk deeltje berekend door numerieke integratie van de interactiepotentiaal en de bewegingswet van Newton. Dit wordt iteratief herhaald, waardoor een simulatietraject30 wordt gegenereerd. Deze berekeningen worden uitgevoerd met een MD-motor; van de verschillende open-sourcepakketten is GROMACS36 een van de meest gebruikte engines en degene die we hier beschrijven. Het bevat ook instrumenten voor het analyseren en construeren van initiële coördinaten van te simuleren systemen37. Andere MD-motoren zijn onder meer NAMD38; CHARMM39 en AMBER40, die de gebruiker naar eigen goeddunken kan selecteren op basis van de rekenkracht van een bepaald systeem. Het is van cruciaal belang om de trajecten tijdens de simulatie te visualiseren, evenals voor analyse en interpretatie van de resultaten. Er zijn verschillende tools beschikbaar; hier bespreken we visuele moleculaire dynamica (VMD) die een breed scala aan functies biedt, waaronder driedimensionale (3D) visualisatie met uitgebreide teken- en kleurmethoden, volumetrische datavisualisatie, het bouwen, voorbereiden en analyseren van trajecten van MD-simulatiesystemen, en het maken van trajectfilms zonder limieten op de systeemgrootte, als het geheugen beschikbaar is41,42,43.
De nauwkeurigheid van de voorspelde dynamiek tussen systeemcomponenten wordt direct beïnvloed door de FF die is gekozen voor de voortplanting van het traject. Empirische FF-parametrisatie-inspanningen worden door weinig onderzoeksgroepen nagestreefd. De meest gevestigde en meest voorkomende FF voor MD zijn CHARMM39, AMBER 40, Martini44, OPLS 45 en SIRAH 46. Het all-atom additive CHARMM36 (C36) krachtveld47 wordt veel gebruikt voor AA MD van membraansystemen, omdat het experimentele structurele gegevens nauwkeurig reproduceert. Het is oorspronkelijk ontwikkeld door de CHARMM-gemeenschap en is compatibel met meerdere MD-engines zoals GROMACS en NAMD. Ondanks verbeteringen in gemeenschappelijke FF’s, is er een voortdurende inspanning om de parametersets te verbeteren om voorspellingen mogelijk te maken die experimentele waarneembare objecten nauwkeurig reproduceren, gedreven door belangen in bepaalde onderzoekssystemen48,49.
Een uitdaging bij het simuleren van lipidemembranen is het bepalen van de lengte van het simulatietraject. Dit is grotendeels afhankelijk van de te analyseren statistieken en het proces dat men wil karakteriseren. Doorgaans hebben complexe lipidenmengsels meer tijd nodig om een evenwicht te bereiken, omdat meer soorten voldoende tijd moeten hebben om op het membraanvlak te diffunderen en een stabiele laterale organisatie te bereiken. Van een simulatie wordt gezegd dat deze in evenwicht is wanneer de eigenschap van belang een plateau heeft bereikt en rond een constante waarde schommelt. Het is gebruikelijk om ten minste 100-200 ns van een evenwichtstraject te verkrijgen om een passende statistische analyse uit te voeren op de eigenschappen en interacties van belang. Het is gebruikelijk om membraansimulaties uit te voeren tussen 200-500 ns, afhankelijk van de complexiteit van het lipidenmengsel en de onderzoeksvraag. Eiwit-lipide-interacties vereisen doorgaans langere simulatietijden, tussen 500-2000 ns. Enkele benaderingen om bemonstering en waarneembare dynamiek met membraansystemen te versnellen zijn: (i) het zeer mobiele membraanmimetische (HMMM)-model, dat eindkoolstofatomen van lipiden in het membraan vervangt door organisch oplosmiddel om bemonstering te versnellen50; en (ii) waterstofmassa-repartitionering (HMR), waarbij een fractie van de massa’s van zware atomen binnen een systeem wordt gecombineerd met die van waterstofatomen om het gebruik van een grotere simulatietijdstap51 mogelijk te maken.
Het volgende protocol bespreekt een beginnersvriendelijke benadering voor het bouwen, uitvoeren en analyseren van realistische membraanmodellen met behulp van AA MD. Gezien de aard van MD-simulaties moeten meerdere trajecten worden uitgevoerd om rekening te houden met reproduceerbaarheid en een goede statistische analyse van de resultaten. Het is momenteel gebruikelijk om minimaal drie replica’s per systeem van belang uit te voeren. Zodra de lipidesoorten zijn geselecteerd voor het organisme en het proces van belang, worden de basisstappen voor het bouwen, uitvoeren en analyseren van een simulatietraject van een membraansysteem geschetst en samengevat in figuur 2.
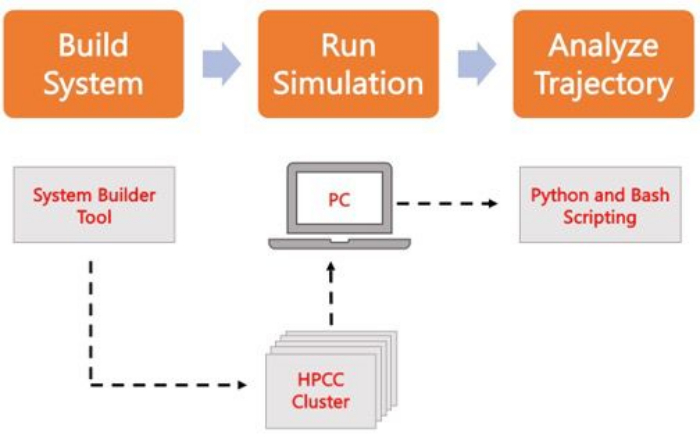
Figuur 2. Schema om MD-simulaties uit te voeren. Oranje vakjes komen overeen met de drie belangrijkste stappen die in het protocol worden beschreven. Daaronder bevindt zich de workflow van het simulatieproces. Tijdens het instellen van het systeem wordt het systeem met de initiële coördinaten van een opgelost membraansysteem gebouwd met een systeeminvoergenerator zoals CHARMM-GUI Membrane Builder. Na het overbrengen van de invoerbestanden naar een high-performance computing-cluster, wordt het simulatietraject gepropageerd met behulp van een MD-engine, zoals GROMACS. Trajectanalyse kan worden uitgevoerd op het computercluster of een lokaal werkstation, samen met visualisatie. Vervolgens wordt de analyse uitgevoerd met behulp van pakketten met ingebouwde analysecode zoals GROMACS en VMD, of met behulp van Bash-scripts of verschillende Python-bibliotheken. Klik hier om een grotere versie van deze figuur te bekijken.
