Les lipides sont des constituants majeurs des membranes, qui fournissent des limites aux cellules et permettent la compartimentation intracellulaire 1,2,3. Les lipides sont amphiphiles, avec un groupe de tête polaire et deux queues d’acides gras hydrophobes ; Ceux-ci s’auto-assemblent en une bicouche pour minimiser le contact des chaînes hydrophobes avec l’eau 3,4. Diverses combinaisons de groupes de têtes hydrophiles et de queues hydrophobes donnent lieu à différentes classes de lipides dans les membranes biologiques, telles que les glycérophospholipides, les sphingolipides et les stérols (Figure 1)1,5,6. Les glycérophospholipides sont les principaux éléments constitutifs des membranes cellulaires eucaryotes composées de glycérophosphate, d’acides gras à longue chaîne et de groupes de tête de faible poids moléculaire7. La nomenclature des lipides est basée sur les différences entre les groupes de têtes ; par exemple, la phosphatidyl-choline (PC), la phosphatidyl-éthanolamine (PE), la phosphatidylsérine (PS), le phosphatidyl-glycérol (PG), le phosphatidyl-inositol (PI) ou l’acide phosphatidique (PA) non modifié5,6. En ce qui concerne les queues hydrophobes, la longueur et le degré de saturation varient, ainsi que la structure de la colonne vertébrale. Les combinaisons possibles sont nombreuses, ce qui donne des milliers d’espèces lipidiques dans les cellules de mammifères6. Les changements dans la composition des lipides membranaires conduisent à différentes propriétés membranaires mécaniques et structurelles qui ont un impact sur l’activité des protéines membranaires intégrales et des protéines périphériques 2,6.
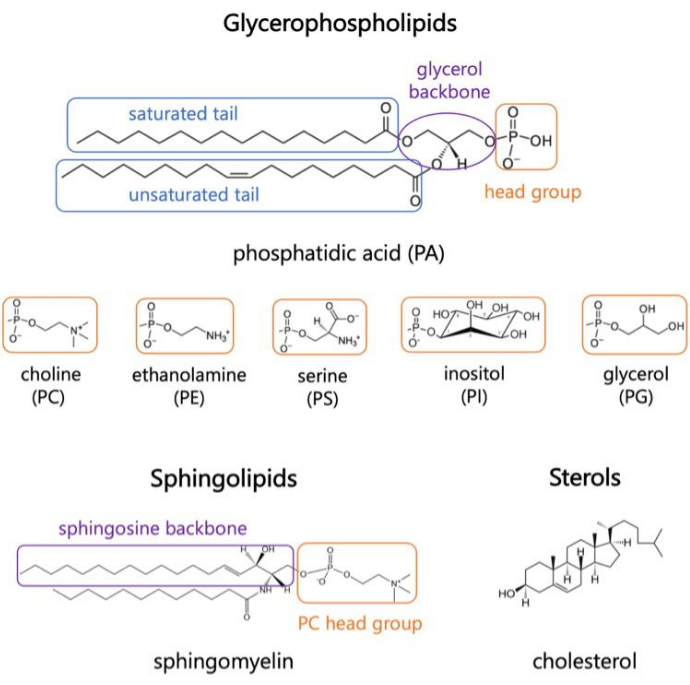
Graphique 1. Structures lipidiques représentatives. Les queues d’acides gras sont représentées dans des cases bleues, les groupes de têtes lipidiques communs en orange et les épines dorsales d’échantillons en violet. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Les lipides jouent un rôle actif dans les processus cellulaires, l’activation des protéines dans les cascades de signalisation et l’homéostasie des cellules saines 8,9. L’altération de la dynamique des lipides est le résultat d’une infection ou peut être un marqueur de pathogenèse de la maladie 10,11,12,13,14,15. En tant que barrières pour la cellule, l’étude des lipides membranaires et de leur rôle dans la perméation de petites molécules est pertinente pour les systèmes d’administration de médicaments et les mécanismes de perturbation membranaire16,17. La diversité chimique et les différents ratios d’espèces lipidiques entre les organites, les tissus et les organismes donnent lieu à une dynamique membranaire complexe2. Il est donc important de conserver ces caractéristiques dans les études de modélisation des bicouches lipidiques, en particulier lorsque l’objectif d’une étude est d’examiner les interactions d’autres biomolécules avec la membrane. Les espèces lipidiques à prendre en compte dans un modèle dépendent de l’organisme et du compartiment cellulaire d’intérêt. Par exemple, les lipides PG sont importants pour le transfert d’électrons dans les bactéries photosynthétiques18, tandis que les lipides d’inositol phosphorylés (PIP) sont des acteurs majeurs de la dynamique de la membrane plasmique (PM) et des cascades de signalisation dans les cellules de mammifères 19,20. À l’intérieur de la cellule, les membranes de PM, de réticulum endoplasmique (RE), de Golgi et mitochondriales contiennent des abondances lipidiques uniques qui influencent leur fonction. Par exemple, le RE est la plaque tournante de la biogenèse des lipides et transporte le cholestérol vers les PM et Golgi ; il contient une grande diversité lipidique avec une abondance de PC et de PE, mais une faible teneur en stérols, ce qui favorise la fluidité membranaire21,22,23,24. En revanche, le PM incorpore des centaines, voire des milliers d’espèces lipidiques selon lesorganismes25, il contient des niveaux élevés de sphingolipides et de cholestérol qui lui confèrent une rigidité caractéristique par rapport aux autres membranes de la cellule24. L’asymétrie des feuillets doit être envisagée pour les membranes comme le PM, qui a un feuillet externe riche en sphingomyéline, en PC et en cholestérol, et un feuillet interne riche en PE, PI et PS qui sont importants pour les cascades de signalisation24. Enfin, la diversité lipidique induit également la formation de micro-domaines qui diffèrent par leur empilement et leur ordre interne, connus sous le nom de radeaux lipidiques24,26 ; Ceux-ci présentent une asymétrie latérale, sont supposés jouer un rôle important dans la signalisation cellulaire26 et sont difficiles à étudier en raison de leur nature transitoire.
Des techniques expérimentales telles que la fluoroscopie, la spectroscopie et des systèmes membranaires modèles tels que les vésicules unilamellaires géantes (GUV) ont été utilisées pour étudier les interactions des biomolécules avec les membranes. Cependant, la nature complexe et dynamique des composants impliqués est difficile à saisir avec les seules méthodes expérimentales. Par exemple, il existe des limites à l’imagerie des domaines transmembranaires des protéines, à la complexité des membranes utilisées dans de telles études et à l’identification d’états intermédiaires ou transitoires au cours du processus d’intérêt27,28,29. Depuis l’avènement de la simulation moléculaire des monocouches et bicouches lipidiques dans les années 198029, les systèmes lipides-protéines et leurs interactions peuvent désormais être quantifiés au niveau moléculaire. La simulation de dynamique moléculaire (MD) est une technique de calcul courante qui prédit le mouvement des particules en fonction de leurs forces intermoléculaires. Un potentiel d’interaction additif décrit les interactions liées et non liées entre les particules du système30. L’ensemble des paramètres utilisés pour modéliser ces interactions est appelé champ de force de simulation (FF). Ces paramètres sont obtenus à partir de calculs ab initio, de calculs semi-empiriques et de mécanique quantique, et optimisés pour reproduire des données provenant d’expériences de diffraction des rayons X et des électrons, de la RMN, de l’infrarouge, de la spectroscopie Raman et de la spectroscopie neutronique, entre autres méthodes31.
Les simulations MD peuvent être utilisées pour étudier des systèmes à différents niveaux de résolution32,33,34. Les systèmes qui visent à caractériser des interactions biomoléculaires spécifiques, des liaisons hydrogène et d’autres détails à haute résolution sont étudiés avec des simulations de tous les atomes (AA). En revanche, les simulations à gros grain (CG) regroupent les atomes en groupes fonctionnels plus grands afin de réduire les coûts de calcul et d’examiner la dynamique à plus grande échelle33. Entre les deux se trouvent des simulations d’atomes unis (UA), où les atomes d’hydrogène sont combinés avec leurs atomes lourds respectifs pour accélérer le calcul33,35. Les simulations MD sont un outil puissant pour l’exploration de la dynamique des membranes lipidiques et de leurs interactions avec d’autres molécules et peuvent servir à fournir des mécanismes au niveau moléculaire pour les processus d’intérêt à l’interface membranaire. De plus, les simulations de DM peuvent servir à affiner les cibles expérimentales et à prédire les propriétés macromoléculaires d’un système donné sur la base d’interactions microscopiques.
En bref, étant donné un ensemble de coordonnées initiales, de vitesses et un ensemble de conditions telles qu’une température et une pression constantes, les positions et les vitesses de chaque particule sont calculées par intégration numérique du potentiel d’interaction et de la loi du mouvement de Newton. Ceci est répété de manière itérative, générant ainsi une trajectoire de simulation30. Ces calculs sont effectués à l’aide d’un moteur MD ; Parmi plusieurs paquets open-source, GROMACS36 est l’un des moteurs les plus couramment utilisés et celui que nous décrivons ici. Il comprend également des outils d’analyse et de construction des coordonnées initiales des systèmes à simuler37. Parmi les autres moteurs MD, citons le NAMD38 ; CHARMM39 et AMBER40, que l’utilisateur peut sélectionner à sa propre discrétion en fonction des performances de calcul d’un système donné. Il est essentiel de visualiser les trajectoires pendant la simulation ainsi que pour l’analyse et l’interprétation des résultats. Une variété d’outils sont disponibles ; Nous discutons ici de la dynamique moléculaire visuelle (VMD) qui offre un large éventail de fonctionnalités, y compris la visualisation tridimensionnelle (3D) avec des méthodes de dessin et de coloration étendues, la visualisation volumétrique des données, la construction, la préparation et l’analyse des trajectoires des systèmes de simulation MD, et la réalisation de films de trajectoire sans limite de taille du système, si la mémoire est disponible41,42,43.
La précision de la dynamique prédite entre les composants du système est directement influencée par le FF choisi pour la propagation de la trajectoire. Les efforts empiriques de paramétrisation de la FF sont poursuivis par quelques groupes de recherche. Les FF les plus établis et les plus courants pour la DM sont CHARMM39, AMBER 40, Martini44, OPLS 45 et SIRAH 46. Le champ de force47 de l’additif CHARMM36 tous les atomes (C36) est largement utilisé pour la MD AA des systèmes membranaires car il reproduit avec précision les données structurelles expérimentales. Il a été développé à l’origine par la communauté CHARMM, et il est compatible avec plusieurs moteurs MD comme GROMACS et NAMD. Malgré les améliorations apportées aux FF courants, il y a un effort continu pour améliorer les ensembles de paramètres afin de permettre des prédictions qui reproduisent fidèlement les observables expérimentales, motivées par l’intérêt pour des systèmes d’étude particuliers48,49.
L’un des défis de la simulation des membranes lipidiques est de déterminer la longueur de la trajectoire de simulation. Cela dépend en grande partie des métriques à analyser et du processus que l’on cherche à caractériser. En règle générale, les mélanges lipidiques complexes nécessitent plus de temps pour atteindre l’équilibre, car un plus grand nombre d’espèces doivent avoir suffisamment de temps pour diffuser sur le plan membranaire et atteindre une organisation latérale stable. On dit d’une simulation qu’elle est à l’équilibre lorsque la propriété d’intérêt a atteint un plateau et fluctue autour d’une valeur constante. Il est courant d’obtenir au moins 100 à 200 ns de trajectoire équilibrée pour effectuer une analyse statistique appropriée sur les propriétés et les interactions d’intérêt. Il est courant d’effectuer des simulations membranaires uniquement entre 200 et 500 ns, en fonction de la complexité du mélange lipidique et de la question de recherche. Les interactions protéine-lipide nécessitent généralement des temps de simulation plus longs, entre 500 et 2000 ns. Voici quelques approches permettant d’accélérer l’échantillonnage et la dynamique observable avec les systèmes membranaires : (i) le modèle mimétique membranaire hautement mobile (HMMM), qui remplace les carbones finaux des lipides dans la membrane par un solvant organique pour accélérer l’échantillonnage50 ; et (ii) le repartitionnement de la masse de l’hydrogène (HMR), qui combine une fraction des masses d’atomes lourds au sein d’un système avec celles des atomes d’hydrogène pour permettre l’utilisation d’un pas de temps de simulation plus grand51.
Le protocole suivant traite d’une approche conviviale pour les débutants afin de créer, d’exécuter et d’analyser des modèles de membrane réalistes à l’aide d’AA MD. Compte tenu de la nature des simulations de DM, plusieurs trajectoires doivent être exécutées pour tenir compte de la reproductibilité et de l’analyse statistique appropriée des résultats. Il est d’usage d’exécuter au moins trois réplicas par système d’intérêt. Une fois que les espèces lipidiques ont été sélectionnées pour l’organisme et le processus d’intérêt, les étapes de base pour construire, exécuter et analyser une trajectoire de simulation d’un système membranaire seul sont décrites et résumées dans la figure 2.
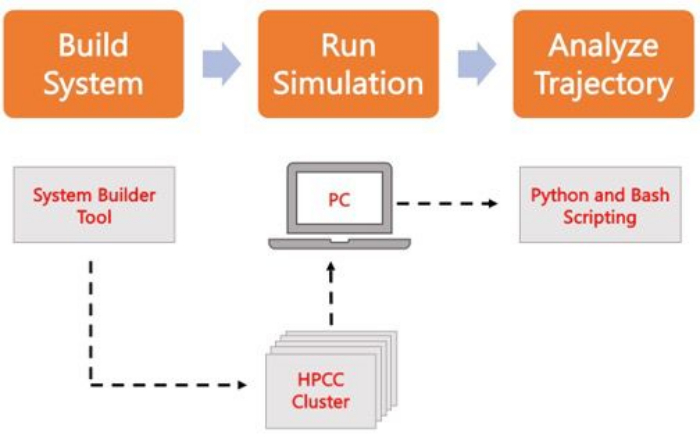
Graphique 2. Schéma permettant d’exécuter des simulations MD. Les cases orange correspondent aux trois étapes principales décrites dans le protocole. En dessous se trouve le flux de travail du processus de simulation. Lors de la configuration du système, le système contenant les coordonnées initiales d’un système à membrane solvatée est construit à l’aide d’un générateur d’entrée système tel que CHARMM-GUI Membrane Builder. Après avoir transféré les fichiers d’entrée vers un cluster de calcul haute performance, la trajectoire de simulation est propagée à l’aide d’un moteur MD, tel que GROMACS. L’analyse de trajectoire peut être effectuée sur le cluster informatique ou sur un poste de travail local avec visualisation. L’analyse est ensuite effectuée, soit à l’aide de packages avec du code d’analyse intégré tels que GROMACS et VMD, soit à l’aide de scripts Bash ou de diverses bibliothèques Python. Veuillez cliquer ici pour voir une version agrandie de cette figure.
