I lipidi sono i principali costituenti delle membrane, che forniscono confini per le cellule e consentono la compartimentazione intracellulare 1,2,3. I lipidi sono anfifilici, con un gruppo di teste polari e due code di acidi grassi idrofobici; Questi si auto-assemblano in un doppio strato per ridurre al minimo il contatto delle catene idrofobiche con l’acqua 3,4. Varie combinazioni di gruppi di teste idrofile e code idrofobiche danno luogo a diverse classi di lipidi nelle membrane biologiche, come glicerofosfosfolipidi, sfingolipidi e steroli (Figura 1)1,5,6. I glicerofosfolipidi sono elementi costitutivi primari delle membrane cellulari eucariotiche composte da glicerofosfato, acidi grassi a catena lunga e gruppi di testa a basso peso molecolare7. La nomenclatura dei lipidi si basa sulle differenze nei gruppi di teste; esempi includono fosfatidil-colina (PC), fosfatidil-etanolammina (PE), fosfatidil-serina (PS), fosfatidil-glicerolo (PG), fosfatidil-inositolo (PI) o acido fosfatidico non modificato (PA)5,6. Per quanto riguarda le code idrofobiche, la lunghezza e il grado di saturazione variano, insieme alla struttura della spina dorsale. Le combinazioni possibili sono numerose, dando vita a migliaia di specie lipidiche nelle cellule dei mammiferi6. I cambiamenti nella composizione lipidica della membrana portano a diverse proprietà meccaniche e strutturali della membrana che influiscono sull’attività sia delle proteine integrali di membrana che delle proteine periferiche 2,6.
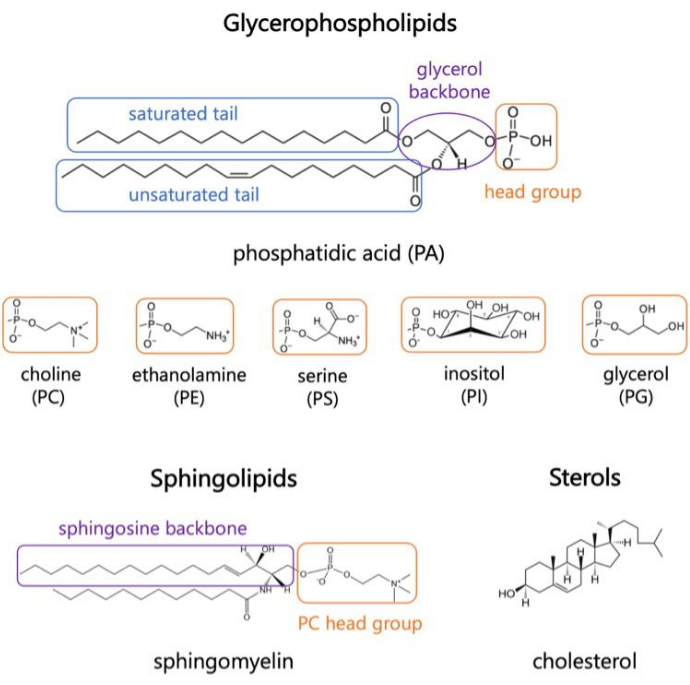
Figura 1. Strutture lipidiche rappresentative. Le code degli acidi grassi sono mostrate in riquadri blu, i gruppi di teste lipidiche comuni in arancione e le spine dorsali dei campioni in viola. Fare clic qui per visualizzare una versione più grande di questa figura.
I lipidi sono attori attivi nei processi cellulari, nell’attivazione delle proteine nelle cascate di segnalazione e nell’omeostasi delle cellule sane 8,9. Le dinamiche lipidiche alterate sono il risultato di un’infezione o possono essere marcatori di patogenesi della malattia 10,11,12,13,14,15. Come barriere per la cellula, lo studio dei lipidi di membrana e del loro ruolo nella permeazione di piccole molecole è di rilevanza per i sistemi di somministrazione dei farmaci e i meccanismi di rottura della membrana16,17. La diversità chimica e i diversi rapporti delle specie lipidiche tra organelli, tessuti e organismi danno origine a complesse dinamiche di membrana2. È quindi importante mantenere queste caratteristiche negli studi di modellizzazione dei doppi strati lipidici, soprattutto quando l’obiettivo di uno studio è quello di esaminare le interazioni di altre biomolecole con la membrana. Le specie lipidiche da considerare in un modello dipendono dall’organismo e dal compartimento cellulare di interesse. Ad esempio, i lipidi PG sono importanti per il trasferimento di elettroni nella bateria fotosintetica18, mentre i lipidi fosforilati dell’inositolo (PIP) sono i principali attori nella dinamica della membrana plasmatica (PM) e nelle cascate di segnalazione nelle cellule di mammifero 19,20. All’interno della cellula, il PM, il reticolo endoplasmatico (ER), il Golgi e le membrane mitocondriali contengono abbondanze lipidiche uniche che influenzano la loro funzione. Ad esempio, l’ER è il fulcro della biogenesi lipidica e trasporta il colesterolo verso il PM e il Golgi; contiene un’elevata diversità lipidica con abbondanza di PC e PE, ma un basso contenuto di steroli, che favorisce la fluidità della membrana21,22,23,24. Al contrario, il PM incorpora centinaia e persino migliaia di specie lipidiche a seconda dell’organismo25, contiene alti livelli di sfingolipidi e colesterolo che gli conferiscono una rigidità caratteristica rispetto ad altre membrane della cellula24. L’asimmetria dei lembi deve essere presa in considerazione per membrane come il PM, che ha un foglietto esterno ricco di sfingomielina, PC e colesterolo e un foglietto interno ricco di PE, PI e PS che sono importanti per le cascate di segnalazione24. Infine, la diversità lipidica induce anche la formazione di micro-domini che differiscono per impacchettamento e ordine interno, noti come zattere lipidiche24,26; Questi mostrano un’asimmetria laterale, si ipotizza che svolgano un ruolo importante nella segnalazione cellulare26 e sono difficili da studiare a causa della loro natura transitoria.
Tecniche sperimentali come la fluoroscopia, la spettroscopia e sistemi di membrane modello come le vescicole unilamellari giganti (GUV) sono state utilizzate per studiare le interazioni delle biomolecole con le membrane. Tuttavia, la natura complessa e dinamica dei componenti coinvolti è difficile da catturare con i soli metodi sperimentali. Ad esempio, ci sono limitazioni sull’imaging dei domini transmembrana delle proteine, sulla complessità delle membrane utilizzate in tali studi e sull’identificazione di stati intermedi o transitori durante il processo di interesse27,28,29. Dall’avvento della simulazione molecolare di monostrati e doppi strati lipidici negli anni ’8029, i sistemi lipidico-proteici e le loro interazioni possono ora essere quantificati a livello molecolare. La simulazione della dinamica molecolare (MD) è una tecnica computazionale comune che prevede il movimento delle particelle in base alle loro forze intermolecolari. Un potenziale di interazione additiva descrive le interazioni legate e non legate tra le particelle del sistema30. L’insieme di parametri utilizzati per modellare queste interazioni è chiamato campo di forza di simulazione (FF). Questi parametri sono ottenuti da calcoli ab initio, calcoli semi-empirici e meccanici quantistici e ottimizzati per riprodurre dati provenienti da esperimenti di diffrazione di raggi X ed elettroni, NMR, infrarossi, Raman e spettroscopia neutronica, tra gli altri metodi31.
Le simulazioni MD possono essere utilizzate per studiare sistemi a vari livelli di risoluzione32,33,34. I sistemi che mirano a caratterizzare specifiche interazioni biomolecolari, legami idrogeno e altri dettagli ad alta risoluzione vengono studiati con simulazioni all-atom (AA). Al contrario, le simulazioni a grana grossa (CG) raggruppano gli atomi in gruppi funzionali più grandi per ridurre i costi computazionali ed esaminare le dinamiche su scala più ampia33. Tra questi due ci sono le simulazioni di atomi uniti (UA), in cui gli atomi di idrogeno sono combinati con i rispettivi atomi pesanti per accelerare il calcolo33,35. Le simulazioni MD sono un potente strumento per l’esplorazione della dinamica delle membrane lipidiche e delle loro interazioni con altre molecole e possono servire a fornire meccanismi a livello molecolare per i processi di interesse all’interfaccia di membrana. Inoltre, le simulazioni MD possono servire a restringere gli obiettivi sperimentali e prevedere le proprietà macromolecolari di un dato sistema sulla base di interazioni microscopiche.
In breve, dato un insieme di coordinate iniziali, velocità e un insieme di condizioni come temperatura e pressione costanti, le posizioni e le velocità di ciascuna particella vengono calcolate attraverso l’integrazione numerica del potenziale di interazione e della legge del moto di Newton. Questa operazione viene ripetuta in modo iterativo, generando così una traiettoria di simulazione30. Questi calcoli vengono eseguiti con un motore MD; tra i vari pacchetti open-source, GROMACS36 è uno dei motori più comunemente usati e quello che descriviamo qui. Include anche strumenti per l’analisi e la costruzione delle coordinate iniziali dei sistemi da simulare37. Altri motori MD includono NAMD38; CHARMM39 e AMBER40, che l’utente può selezionare a propria discrezione in base alle prestazioni computazionali di un determinato sistema. È fondamentale visualizzare le traiettorie durante la simulazione, nonché per l’analisi e l’interpretazione dei risultati. Sono disponibili una varietà di strumenti; qui discutiamo la dinamica molecolare visiva (VMD) che offre un’ampia gamma di funzionalità, tra cui la visualizzazione tridimensionale (3-D) con metodi di disegno e colorazione espansivi, la visualizzazione dei dati volumetrici, la costruzione, la preparazione e l’analisi delle traiettorie dei sistemi di simulazione MD e la creazione di filmati di traiettorie senza limiti sulle dimensioni del sistema, se la memoria è disponibile41,42,43.
L’accuratezza della dinamica prevista tra i componenti del sistema è direttamente influenzata dal FF scelto per la propagazione della traiettoria. Gli sforzi di parametrizzazione empirica di FF sono perseguiti da pochi gruppi di ricerca. I FF più consolidati e comuni per MD includono CHARMM39, AMBER 40, Martini44, OPLS 45 e SIRAH 46. Il campo di forza47 dell’additivo CHARMM36 di tutti gli atomi (C36) è ampiamente utilizzato per l’AA MD dei sistemi a membrana, in quanto riproduce accuratamente i dati strutturali sperimentali. È stato originariamente sviluppato dalla comunità CHARMM ed è compatibile con più motori MD come GROMACS e NAMD. Nonostante i miglioramenti tra i FF comuni, c’è uno sforzo continuo per migliorare i set di parametri per consentire previsioni che riproducano fedelmente le osservabili sperimentali, guidato dall’interesse per particolari sistemi di studio48,49.
Una sfida quando si simulano le membrane lipidiche è determinare la lunghezza della traiettoria di simulazione. Ciò dipende in gran parte dalle metriche da analizzare e dal processo che si intende caratterizzare. Tipicamente, le miscele lipidiche complesse richiedono più tempo per raggiungere l’equilibrio, poiché più specie devono avere abbastanza tempo per diffondersi sul piano della membrana e raggiungere un’organizzazione laterale stabile. Si dice che una simulazione è in equilibrio quando la proprietà di interesse ha raggiunto un plateau e fluttua intorno a un valore costante. E’ pratica comune ottenere almeno 100-200 ns di traiettoria equilibrata per effettuare opportune analisi statistiche sulle proprietà e interazioni di interesse. È comune eseguire simulazioni di sola membrana tra 200-500 ns, a seconda della complessità della miscela lipidica e della domanda di ricerca. Le interazioni proteina-lipidi richiedono in genere tempi di simulazione più lunghi, tra 500 e 2000 ns. Alcuni approcci per accelerare il campionamento e le dinamiche osservabili con i sistemi a membrana sono: (i) il modello mimetico di membrana altamente mobile (HMMM), che sostituisce i carboni finali dei lipidi nella membrana con solvente organico per accelerare il campionamento50; e (ii) il ripartizionamento della massa dell’idrogeno (HMR), che combina una frazione delle masse degli atomi pesanti all’interno di un sistema con quelle degli atomi di idrogeno per consentire l’uso di una simulazione più ampia del timestep51.
Il seguente protocollo illustra un approccio adatto ai principianti per costruire, eseguire e analizzare modelli di membrana realistici utilizzando AA MD. Data la natura delle simulazioni MD, è necessario eseguire più traiettorie per tenere conto della riproducibilità e di una corretta analisi statistica dei risultati. La prassi corrente prevede l’esecuzione di almeno tre repliche per ogni sistema di interesse. Una volta che le specie lipidiche sono state selezionate per l’organismo e il processo di interesse, i passaggi di base per costruire, eseguire e analizzare una traiettoria di simulazione di un sistema di sola membrana sono delineati e riassunti nella Figura 2.
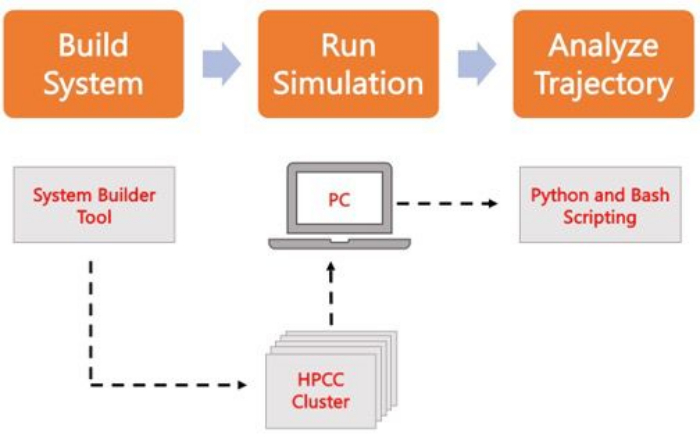
Figura 2. Schema per l’esecuzione di simulazioni MD. Le caselle arancioni corrispondono alle tre fasi principali descritte nel protocollo. Di seguito è riportato il flusso di lavoro del processo di simulazione. Durante la configurazione del sistema, il sistema contenente le coordinate iniziali di un sistema a membrana solvatato viene costruito con un generatore di input di sistema come CHARMM-GUI Membrane Builder. Dopo aver trasferito i file di input in un cluster di calcolo ad alte prestazioni, la traiettoria di simulazione viene propagata utilizzando un motore MD, ad esempio GROMACS. L’analisi della traiettoria può essere eseguita sul cluster di computer o su una stazione di lavoro locale insieme alla visualizzazione. L’analisi viene quindi eseguita, utilizzando pacchetti con codice di analisi incorporato come GROMACS e VMD, oppure utilizzando script Bash o varie librerie Python. Fare clic qui per visualizzare una versione più grande di questa figura.
