脂質は膜の主要成分であり、細胞の境界を提供し、細胞内の区画化を可能にします1,2,3。脂質は両親媒性で、極性頭部群と2つの疎水性脂肪酸尾部があります。これらは自己組織化して二重層になり、疎水性鎖と水との接触が最小限に抑えられます3,4。親水性の頭部基と疎水性の尾部のさまざまな組み合わせにより、グリセロリン脂質、スフィンゴ脂質、ステロールなど、生体膜にさまざまなクラスの脂質が生じます(図1)1,5,6。グリセロリン脂質は、グリセロリン酸、長鎖脂肪酸、および低分子量の頭部基で構成される真核細胞膜の主要な構成要素です7。脂質の命名法は、ヘッドグループの違いに基づいています。例としては、ホスファチジルコリン(PC)、ホスファチジルエタノールアミン(PE)、ホスファチジルセリン(PS)、ホスファチジルグリセロール(PG)、ホスファチジルイノシトール(PI)、または非修飾ホスファチジン酸(PA)などがあります5,6。疎水性の尾部に関しては、背骨構造とともに、飽和の長さと程度が異なります。可能な組み合わせは多数あり、その結果、哺乳類細胞には数千の脂質種が生じます6。膜脂質組成の変化は、内在性膜タンパク質と末梢タンパク質の両方の活性に影響を与える異なる機械的および構造的膜特性につながります2,6。
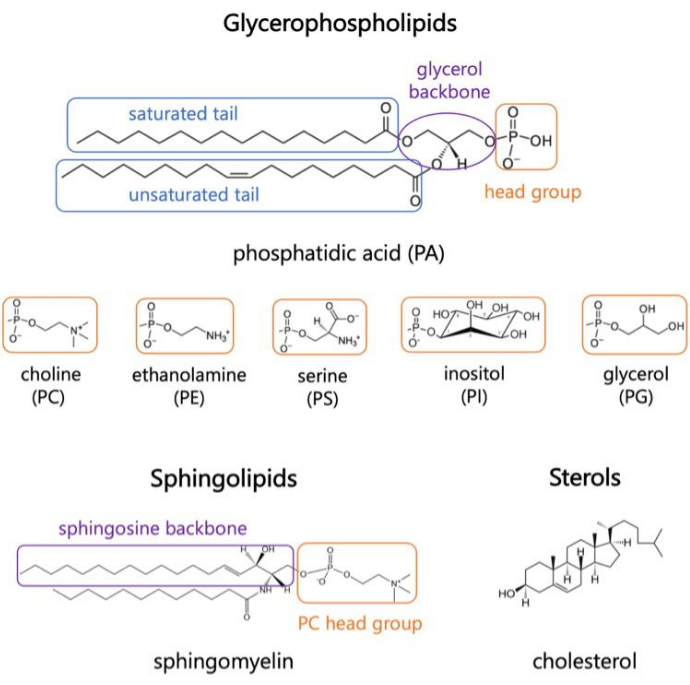
図 1.代表的な脂質構造。 脂肪酸のテールは青色のボックスで、一般的な脂質ヘッドグループはオレンジ色で、サンプルの骨格は紫色で示されています。 この図の拡大版をご覧になるには、ここをクリックしてください。
脂質は、細胞プロセス、シグナル伝達カスケードにおけるタンパク質の活性化、および健康な細胞の恒常性における積極的なプレーヤーです8,9。脂質動態の変化は、感染の結果であるか、疾患の病因のマーカーである可能性があります10,11,12,13,14,15。細胞のバリアとして、膜脂質と低分子の浸透におけるそれらの役割の研究は、薬物送達システムと膜破壊メカニズムに関連しています16,17。細胞小器官、組織、生物における化学的多様性と脂質種の比率の違いは、複雑な膜ダイナミクスを引き起こします2。したがって、脂質二重層のモデリング研究において、特に研究の目的が他の生体分子と膜との相互作用を調べることである場合、これらの特性を保持することが重要です。モデルで考慮する脂質種は、対象となる生物と細胞区画によって異なります。例えば、PG脂質は光合成バテリアの電子移動に重要であり18、リン酸化イノシトール脂質(PIP)は哺乳類細胞の原形質膜(PM)の動態とシグナル伝達カスケードの主要な役割を担っている19,20。細胞内では、PM、小胞体(ER)、ゴルジ体、およびミトコンドリア膜には、その機能に影響を与える固有の脂質量が含まれています。例えば、小胞体は脂質の生合成のハブであり、コレステロールをPMとゴルジ体に輸送します。脂質の多様性が高く、PCとPEが豊富に含まれていますが、ステロール含有量は低く、膜の流動性を促進します21,22,23,24。対照的に、PMは、生物25に応じて数百、さらには数千の脂質種を組み込んでおり、高レベルのスフィンゴ脂質およびコレステロールを含み、細胞内の他の膜と比較して特徴的な剛性を与える24。スフィンゴミエリン、PC、コレステロールが豊富な外側のリーフレットと、シグナル伝達カスケードに重要なPE、PI、およびPSが豊富な内側リーフレットを持つPMのような膜では、リーフレットの非対称性を考慮する必要があります24。最後に、脂質の多様性は、脂質ラフトとして知られる、パッキングと内部秩序が異なるミクロドメインの形成も促します24,26。これらは横方向の非対称性を示し、細胞シグナル伝達に重要な役割を果たすと仮定されており26、その一過性の性質のために研究が困難である。
蛍光透視法、分光法、巨大単層小胞(GUV)などのモデル膜系などの実験技術を使用して、生体分子と膜の相互作用が研究されてきました。しかし、関与する成分の複雑で動的な性質は、実験的手法だけでは捉えることが困難です。例えば、タンパク質の膜貫通ドメインのイメージング、そのような研究で使用される膜の複雑さ、および関心のあるプロセス中の中間状態または過渡状態の同定には制限があります27,28,29。1980年代に脂質単層および二重膜の分子シミュレーションが登場して以来29、脂質-タンパク質系とその相互作用を分子レベルで定量化できるようになりました。分子動力学(MD)シミュレーションは、分子間力に基づいて粒子の動きを予測する一般的な計算手法です。相加的相互作用ポテンシャルは、系30の粒子間の結合および非結合の相互作用を記述する。これらの相互作用をモデル化するために使用される一連のパラメータは、シミュレーション力場(FF)と呼ばれます。これらのパラメータは、第一原理計算、半経験的計算、量子力学的計算から得られ、X線および電子回折実験、NMR、赤外線、ラマンおよび中性子分光法などの方法から再現されたデータに最適化されています31。
MDシミュレーションは、さまざまなレベルの分解能32、33、34でシステムを研究するために使用できます。特定の生体分子相互作用、水素結合、その他の高分解能の詳細を特徴付けることを目的としたシステムは、全原子(AA)シミュレーションで研究されています。対照的に、粗視化(CG)シミュレーションは、計算コストを削減し、より大きなスケールのダイナミクスを調べるために、原子をより大きな官能基にまとめる33。これら2つの間に位置するのは、水素原子がそれぞれの重原子と組み合わされて計算を高速化する結合原子(UA)シミュレーションです33,35。MDシミュレーションは、脂質膜のダイナミクスと他の分子との相互作用を探索するための強力なツールであり、膜界面で関心のあるプロセスに分子レベルのメカニズムを提供するのに役立ちます。さらに、MDシミュレーションは、実験ターゲットを絞り込み、微視的相互作用に基づいて特定のシステムの高分子特性を予測するのに役立ちます。
簡単に言うと、初期座標、速度、温度や圧力などの条件が一定であれば、相互作用ポテンシャルとニュートンの運動の法則を数値積分して、各粒子の位置と速度が計算されます。これを反復的に繰り返すことで、シミュレーション軌跡30を生成する。これらの計算は MD エンジンで実行されます。いくつかのオープンソースパッケージの中で、GROMACS36は最も一般的に使用されているエンジンの1つであり、ここで説明するエンジンです。また、シミュレーションするシステムの初期座標を解析および構築するためのツールも含まれています37。他のMDエンジンにはNAMD38が含まれます。CHARMM39、およびAMBER40は、ユーザが所与のシステムの計算性能に基づいて自身の裁量で選択することができる。シミュレーション中の軌跡を視覚化し、結果を解析および解釈することが重要です。さまざまなツールが利用可能です。ここでは、拡張的な描画法や色付け法による3次元(3D)可視化、体積データの可視化、MDシミュレーションシステムの構築・準備・解析、メモリに余裕があればシステムサイズに制限のない軌道動画作成など、幅広い機能を提供するVisual Molecular Dynamics(VMD)について述べる41,42,43。
システムコンポーネント間の予測ダイナミクスの精度は、軌跡の伝搬に選択されたフリップフロップに直接影響されます。経験的なFFパラメータ化の取り組みは、少数の研究グループによって追求されています。MDの最も確立され、一般的なFFには、CHARMM39、AMBER 40、Martini44、OPLS 45、およびSIRAH 46が含まれます。全原子添加CHARMM36(C36)力場47は、実験構造データを正確に再現するため、膜系のAA MDに広く用いられている。もともとはCHARMMコミュニティによって開発されたもので、GROMACSやNAMDなどの複数のMDエンジンと互換性があります。一般的なFF全体の改善にもかかわらず、特定の研究システムへの関心に駆り立てられて、実験観測結果を厳密に再現する予測を可能にするためにパラメータセットを改善するための継続的な努力があります48,49。
脂質膜をシミュレーションする際の課題は、シミュレーションの軌跡の長さを決定することです。これは、分析する指標と特徴付けるプロセスに大きく依存します。通常、複雑な脂質混合物は、より多くの分子種が膜面上で拡散し、安定した側方組織に到達するのに十分な時間を必要とするため、平衡に達するまでに長い時間を必要とします。シミュレーションは、対象のプロパティがプラトーに達し、一定の値を中心に変動する場合に平衡状態にあると言われます。少なくとも100〜200 nsの平衡軌道を取得して、関心のある特性と相互作用に関する適切な統計分析を実行するのが一般的です。200〜500 nsの間でメンブレンのみのシミュレーションを実行するのが一般的ですが、脂質混合物の複雑さと研究課題によって異なります。タンパク質と脂質の相互作用は、通常、500〜2000nsの長いシミュレーション時間を必要とします。メンブレンシステムによるサンプリングと観察可能なダイナミクスを加速するためのいくつかのアプローチは、(i)膜内の脂質の末端炭素を有機溶媒に置き換えてサンプリングを加速する高移動性膜模倣(HMMM)モデルです50;(ii)水素質量再分配(HMR)は、システム内の重原子の質量の一部を水素原子の質量と組み合わせて、より大きなシミュレーションタイムステップ51の使用を可能にする。
以下のプロトコルでは、AA MD を使用して現実的な膜モデルを構築、実行、および解析するための初心者向けのアプローチについて説明します。MDシミュレーションの性質上、再現性と結果の適切な統計解析を考慮するために、複数の軌跡を実行する必要があります。現在のところ、対象のシステムごとに少なくとも 3 つのレプリカを実行するのが一般的です。目的の生物種とプロセスに脂質種を選択したら、膜のみのシステムのシミュレーション軌跡を構築、実行、および分析するための基本的な手順を概説し、 図 2 にまとめます。
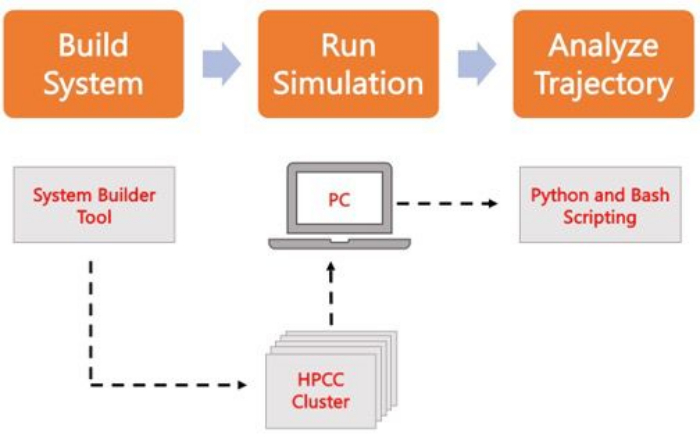
図 2.MDシミュレーションを実行するための回路図。 オレンジ色のボックスは、プロトコルに記載されている 3 つの主要なステップに対応しています。その下には、シミュレーションプロセスのワークフローがあります。システムのセットアップ中に、溶媒和膜システムの初期座標を含むシステムが、CHARMM-GUI Membrane Builderなどのシステム入力ジェネレータを使用して構築されます。入力ファイルをハイパフォーマンスコンピューティングクラスタに転送した後、GROMACSなどのMDエンジンを使用してシミュレーションの軌跡を伝搬します。軌道解析は、コンピュータクラスタまたはローカルワークステーション上で、視覚化とともに行うことができます。その後、GROMACSやVMDなどの解析コードが組み込まれたパッケージ、またはBashスクリプトや各種Pythonライブラリを使用して解析を行います。 この図の拡大版をご覧になるには、ここをクリックしてください。
