Os lipídios são os principais constituintes das membranas, que fornecem limites para as células e permitem a compartimentalização intracelular 1,2,3. Os lipídios são anfifílicos, com um grupo de cabeça polar e duas caudas de ácidos graxos hidrofóbicos; Estes se auto-agrupam em uma bicamada para minimizar o contato das cadeias hidrofóbicas com a água 3,4. Várias combinações de grupos cabeçais hidrofílicos e caudas hidrofóbicas resultam em diferentes classes de lipídios nas membranas biológicas, como glicerofosfolipídios, esfingolipídios e esteróis (Figura 1)1,5,6. Os glicerofosfolipídios são blocos de construção primários das membranas celulares eucarióticas compostas por glicerofosfato, ácidos graxos de cadeia longa e grupos de cabeça de baixo peso molecular7. A nomenclatura lipídica é baseada em diferenças nos grupos de cabeça; exemplos incluem fosfatidil-colina (PC), fosfatidil-etanolamina (PE), fosfatidil-serina (PS), fosfatidil-glicerol (PG), fosfatidil-inositol (PI) ou ácido fosfatídico (PA) não modificado5,6. Quanto às caudas hidrofóbicas, o comprimento e o grau de saturação variam, juntamente com a estrutura da espinha dorsal. As combinações possíveis são inúmeras, resultando em milhares de espécies lipídicas em células demamíferos6. Alterações na composição lipídica da membrana levam a diferentes propriedades mecânicas e estruturais da membrana que afetam a atividade tanto das proteínas integrais de membrana quanto das proteínas periféricas 2,6.
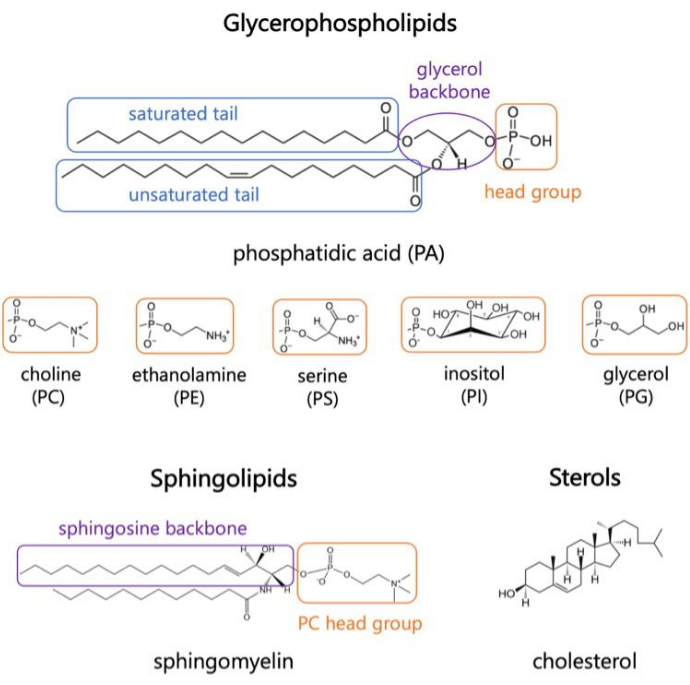
Gráfico 1. Estruturas lipídicas representativas. Caudas de ácidos graxos são mostradas em caixas azuis, grupos de cabeças lipídicas comuns em laranja e espinhas dorsais de amostra em roxo. Clique aqui para ver uma versão maior desta figura.
Os lipídios são atores ativos nos processos celulares, na ativação de proteínas em cascatas de sinalização e na homeostase de células saudáveis 8,9. Alterações na dinâmica lipídica são resultado de infecção ou podem ser marcadores de patogênese da doença10,11,12,13,14,15. Como barreiras para a célula, o estudo dos lipídios da membrana e seu papel na permeação de pequenas moléculas é de relevância para os sistemas de liberação de fármacos e mecanismos de ruptura de membrana16,17. A diversidade química e as diferentes proporções de espécies lipídicas entre organelas, tecidos e organismos dão origem a uma complexa dinâmica de membranas2. Portanto, é importante manter essas características em estudos de modelagem de bicamadas lipídicas, especialmente quando o objetivo de um estudo é examinar as interações de outras biomoléculas com a membrana. As espécies lipídicas a serem consideradas em um modelo dependem do organismo e do compartimento celular de interesse. Por exemplo, os lipídios PG são importantes para a transferência de elétrons na bateria fotossintética18, enquanto os lipídios inositol fosforilados (PIPs) são os principais agentes na dinâmica da membrana plasmática (PM) e cascatas de sinalização em células de mamíferos19,20. Dentro da célula, as membranas PM, retículo endoplasmático (RE), Golgi e mitocondrial contêm abundâncias lipídicas únicas que influenciam sua função. Por exemplo, o ER é o centro da biogênese lipídica e transporta o colesterol para o PM e Golgi; contém alta diversidade lipídica com abundância de PC e PE, mas baixo teor de esteróis, o que promove fluidez da membrana21,22,23,24. Em contrapartida, o MP incorpora centenas e até milhares de espécies lipídicas dependendo do organismo25, contém altos níveis de esfingolipídios e colesterol que lhe conferem rigidez característica em relação a outras membranas da célula24. A assimetria da cúspide deve ser considerada para membranas como a PM, que possui uma cúspide externa rica em esfingomielina, PC e colesterol, e uma cúspide interna rica em EP, PI e SP, importantes para a sinalização de cascatas24. Finalmente, a diversidade lipídica também propicia a formação de microdomínios que diferem em empacotamento e ordem interna, conhecidos como jangadas lipídicas24,26; Estes exibem assimetria lateral, são hipotetizados para desempenhar papéis importantes na sinalização celular26, e são difíceis de estudar devido à sua natureza transitória.
Técnicas experimentais como fluoroscopia, espectroscopia e sistemas de membrana modelo como vesículas unilamelares gigantes (GUVs) têm sido usadas para investigar interações de biomoléculas com membranas. No entanto, a natureza complexa e dinâmica dos componentes envolvidos é difícil de capturar apenas com métodos experimentais. Por exemplo, existem limitações na obtenção de imagens de domínios transmembrana de proteínas, na complexidade das membranas utilizadas nesses estudos e na identificação de estados intermediários ou transitórios durante o processo de interesse27,28,29. Desde o advento da simulação molecular de monocamadas e bicamadas lipídicas na década de 198029, os sistemas lipídico-proteicos e suas interações podem agora ser quantificados em nível molecular. A simulação de dinâmica molecular (DM) é uma técnica computacional comum que prevê o movimento de partículas com base em suas forças intermoleculares. Um potencial de interação aditivo descreve as interações ligadas e não ligadas entre partículas do sistema30. O conjunto de parâmetros usados para modelar essas interações é denominado campo de força de simulação (FF). Esses parâmetros são obtidos a partir de cálculos ab initio, semi-empíricos e de mecânica quântica, e otimizados para reproduzir dados de experimentos de difração de raios X e elétrons, RMN, infravermelho, espectroscopia Raman e de nêutrons, entre outros métodos31.
Simulações MD podem ser usadas para estudar sistemas em vários níveis de resolução32,33,34. Sistemas que visam caracterizar interações biomoleculares específicas, ligações de hidrogênio e outros detalhes de alta resolução são estudados com simulações de átomos (AA). Em contraste, simulações de grão grosso (CG) agrupam átomos em grupos funcionais maiores para reduzir o custo computacional e examinar a dinâmica em maior escala33. Entre esses dois estão as simulações de átomos unidos (UA), onde átomos de hidrogênio são combinados com seus respectivos átomos pesados para acelerar a computação33,35. Simulações MD são uma ferramenta poderosa para a exploração da dinâmica de membranas lipídicas e suas interações com outras moléculas e podem servir para fornecer mecanismos de nível molecular para processos de interesse na interface membrana. Adicionalmente, simulações MD podem servir para estreitar alvos experimentais e prever propriedades macromoleculares de um dado sistema com base em interações microscópicas.
Em resumo, dado um conjunto de coordenadas iniciais, velocidades e um conjunto de condições como temperatura e pressão constantes, as posições e velocidades de cada partícula são calculadas através da integração numérica do potencial de interação e da Lei do movimento de Newton. Isso se repete iterativamente, gerando uma trajetória de simulação30. Esses cálculos são realizados com um motor MD; entre vários pacotes de código aberto, o GROMACS36 é um dos mecanismos mais usados e o que descrevemos aqui. Inclui também ferramentas para análise e construção de coordenadas iniciais de sistemas a serem simulados37. Outros motores MD incluem NAMD38; CHARMM39 e AMBER40, que o usuário pode selecionar a seu próprio critério com base no desempenho computacional de um determinado sistema. É fundamental visualizar as trajetórias durante a simulação, bem como para análise e interpretação dos resultados. Uma variedade de ferramentas estão disponíveis; aqui discutimos a dinâmica molecular visual (VMD) que oferece uma ampla gama de recursos, incluindo visualização tridimensional (3D) com métodos expansivos de desenho e coloração, visualização volumétrica de dados, construção, preparação e análise de trajetórias de sistemas de simulação MD e produção de trajetória-filme sem limites no tamanho do sistema, se a memória estiver disponível41,42,43.
A precisão da dinâmica prevista entre os componentes do sistema é diretamente influenciada pelo FF escolhido para a propagação da trajetória. Os esforços empíricos de parametrização dos FF são perseguidos por poucos grupos de pesquisa. Os FF mais estabelecidos e comuns para MD incluem CHARMM39, AMBER 40, Martini44, OPLS 45 e SIRAH 46. O campo de força de CHARMM36 aditivo de todos os átomos (C36)47 é amplamente utilizado para DM AA de sistemas de membrana, pois reproduz com precisão dados estruturais experimentais. Foi originalmente desenvolvido pela comunidade CHARMM, e é compatível com vários motores MD como GROMACS e NAMD. Apesar das melhorias entre os FFs comuns, há um esforço contínuo para melhorar os conjuntos de parâmetros para permitir previsões que reproduzam de perto observáveis experimentais, impulsionados por interesses em sistemas particulares de estudo48,49.
Um desafio ao simular membranas lipídicas é determinar o comprimento da trajetória da simulação. Isso depende muito das métricas a serem analisadas e do processo que se pretende caracterizar. Tipicamente, misturas lipídicas complexas requerem maior tempo para atingir o equilíbrio, pois mais espécies devem ter tempo suficiente para se difundir no plano da membrana e alcançar uma organização lateral estável. Uma simulação é dita estar em equilíbrio quando a propriedade de interesse atingiu um platô e flutua em torno de um valor constante. É prática comum obter pelo menos 100-200 ns de trajetória equilibrada para realizar análises estatísticas apropriadas sobre as propriedades e interações de interesse. É comum executar simulações somente de membrana entre 200-500 ns, dependendo da complexidade da mistura lipídica e da questão de pesquisa. Interações proteína-lipídio tipicamente requerem tempos de simulação mais longos, entre 500-2000 ns. Algumas abordagens para acelerar a amostragem e a dinâmica observável com sistemas de membrana são: (i) o modelo mimético de membrana altamente móvel (HMMM), que substitui carbonos finais de lipídios na membrana por solvente orgânico para acelerar a amostragem50; e (ii) reparticionamento de massa de hidrogênio (HMR), que combina uma fração das massas de átomos pesados dentro de um sistema com as de átomos de hidrogênio para permitir o uso de um maior tempo de simulação51.
O protocolo a seguir discute uma abordagem amigável para iniciantes para construir, executar e analisar modelos de membrana realistas usando AA MD. Dada a natureza das simulações MD, múltiplas trajetórias devem ser executadas para dar conta da reprodutibilidade e da análise estatística adequada dos resultados. É prática corrente executar no mínimo três réplicas por sistema de interesse. Uma vez que as espécies lipídicas tenham sido selecionadas para o organismo e processo de interesse, as etapas básicas para construir, executar e analisar uma trajetória de simulação de um sistema somente de membrana são descritas e resumidas na Figura 2.
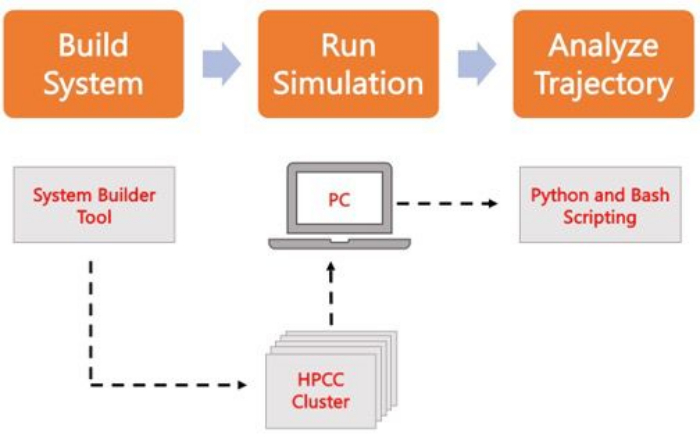
Gráfico 2. Esquema para executar simulações MD. As caixas laranja correspondem às três etapas principais descritas no protocolo. Abaixo está o fluxo de trabalho do processo de simulação. Durante a configuração do sistema, o sistema que contém as coordenadas iniciais de um sistema de membrana solvatada é construído com um gerador de entrada do sistema como o CHARMM-GUI Membrane Builder. Depois de transferir os arquivos de entrada para um cluster de computação de alto desempenho, a trajetória de simulação é propagada usando um mecanismo MD, como o GROMACS. A análise de trajetória pode ser feita no cluster de computadores ou em uma estação de trabalho local, juntamente com a visualização. A análise é então realizada, usando pacotes com código de análise interno, como GROMACS e VMD, ou usando scripts Bash ou várias bibliotecas Python. Clique aqui para ver uma versão maior desta figura.
