Los lípidos son los principales constituyentes de las membranas, que proporcionan límites para las células y permiten la compartimentación intracelular 1,2,3. Los lípidos son anfifílicos, con un grupo de cabeza polar y dos colas de ácidos grasos hidrofóbicos; Estos se autoensamblan en una bicapa para minimizar el contacto de las cadenas hidrofóbicas con el agua 3,4. Varias combinaciones de grupos de cabezas hidrofílicas y colas hidrofóbicas dan como resultado diferentes clases de lípidos en las membranas biológicas, como glicerofosfolípidos, esfingolípidos y esteroles (Figura 1)1,5,6. Los glicerofosfolípidos son componentes primarios de las membranas celulares eucariotas compuestas de glicerofosfato, ácidos grasos de cadena larga y grupos principales de bajo peso molecular7. La nomenclatura lipídica se basa en las diferencias en los grupos de cabeza; algunos ejemplos son la fosfatidilcolina (PC), la fosfatidil-etanolamina (PE), la fosfatidil-serina (PS), el fosfatidilglicerol (PG), el fosfatidil-inositol (PI) o el ácido fosfatídico (PA) no modificado5,6. En cuanto a las colas hidrofóbicas, la longitud y el grado de saturación varían, junto con la estructura de la columna vertebral. Las combinaciones posibles son numerosas, resultando en miles de especies lipídicas en células de mamíferos6. Los cambios en la composición lipídica de la membrana conducen a diferentes propiedades mecánicas y estructurales de la membrana que afectan la actividad tanto de las proteínas integrales de membrana como de las proteínas periféricas 2,6.
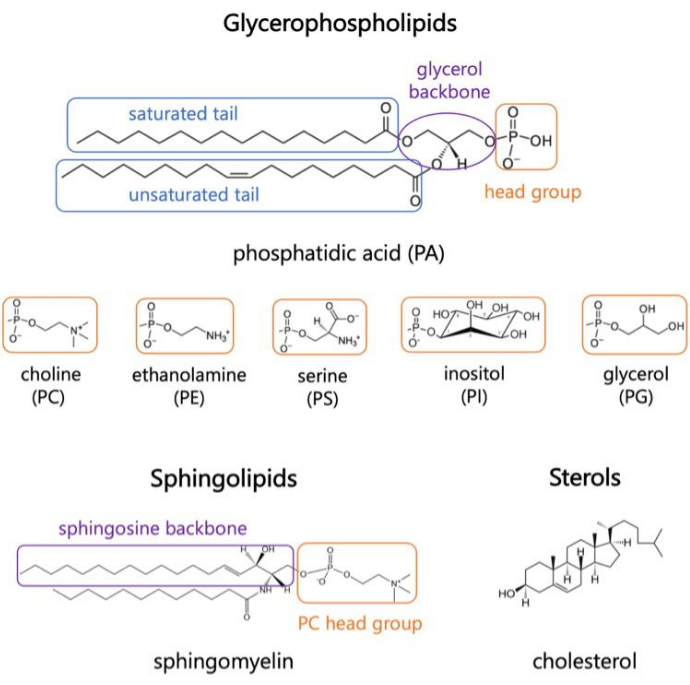
Figura 1. Estructuras lipídicas representativas. Las colas de ácidos grasos se muestran en cuadros azules, los grupos de cabezas de lípidos comunes en naranja y las columnas vertebrales de las muestras en púrpura. Haga clic aquí para ver una versión más grande de esta figura.
Los lípidos son actores activos en los procesos celulares, en la activación de proteínas en las cascadas de señalización y en la homeostasis de las células sanas 8,9. La alteración de la dinámica lipídica es el resultado de la infección o puede ser marcador de patogenia de la enfermedad 10,11,12,13,14,15. Como barreras para la célula, el estudio de los lípidos de membrana y su papel en la permeación de moléculas pequeñas es relevante para los sistemas de liberación de fármacos y los mecanismos de disrupción de la membrana16,17. La diversidad química y las diferentes proporciones de especies lipídicas en orgánulos, tejidos y organismos dan lugar a una dinámica compleja de membranas2. Por lo tanto, es importante mantener estas características en los estudios de modelización de bicapas lipídicas, especialmente cuando el objetivo de un estudio es examinar las interacciones de otras biomoléculas con la membrana. Las especies lipídicas a considerar en un modelo dependen del organismo y del compartimento celular de interés. Por ejemplo, los lípidos PG son importantes para la transferencia de electrones en la batería fotosintética18, mientras que los lípidos fosforilados de inositol (PIP) son actores importantes en la dinámica de la membrana plasmática (PM) y en las cascadas de señalización en las células de mamíferos19,20. Dentro de la célula, el PM, el retículo endoplásmico (RE), Golgi y las membranas mitocondriales contienen abundancias lipídicas únicas que influyen en su función. Por ejemplo, el RE es el centro de la biogénesis lipídica y transporta el colesterol hacia el PM y el Golgi; contiene una alta diversidad lipídica con abundancia de PC y PE, pero bajo contenido de esteroles, lo que promueve la fluidez de la membrana21,22,23,24. Por el contrario, el PM incorpora cientos e incluso miles de especies lipídicas dependiendo del organismo25, contiene altos niveles de esfingolípidos y colesterol que le confieren una rigidez característica en comparación con otras membranas de la célula24. Se debe considerar la asimetría de las valvas para membranas como la PM, que tiene una valva externa rica en esfingomielina, PC y colesterol, y una valva interna rica en PE, PI y PS que son importantes para las cascadas de señalización24. Finalmente, la diversidad lipídica también propicia la formación de microdominios que difieren en el empaquetamiento y el orden interno, conocidos como balsas lipídicas24,26; Estos exhiben asimetría lateral, se supone que juegan un papel importante en la señalización celular26 y son difíciles de estudiar debido a su naturaleza transitoria.
Se han utilizado técnicas experimentales como la fluoroscopia, la espectroscopia y sistemas de membranas modelo como las vesículas unilamelares gigantes (GUV) para investigar las interacciones de las biomoléculas con las membranas. Sin embargo, la naturaleza compleja y dinámica de los componentes involucrados es difícil de capturar solo con métodos experimentales. Por ejemplo, existen limitaciones en la obtención de imágenes de los dominios transmembrana de las proteínas, la complejidad de las membranas utilizadas en dichos estudios y la identificación de estados intermedios o transitorios durante el proceso de interés27,28,29. Desde el advenimiento de la simulación molecular de monocapas y bicapas lipídicas en la década de 198029, los sistemas lípido-proteína y sus interacciones pueden cuantificarse a nivel molecular. La simulación de dinámica molecular (MD) es una técnica computacional común que predice el movimiento de las partículas en función de sus fuerzas intermoleculares. Un potencial de interacción aditiva describe las interacciones enlazadas y no enlazadas entre las partículas del sistema30. El conjunto de parámetros utilizados para modelar estas interacciones se denomina campo de fuerza de simulación (FF). Estos parámetros se obtienen a partir de cálculos ab initio, semiempíricos y de mecánica cuántica, y se optimizan para reproducir datos de experimentos de rayos X y difracción de electrones, RMN, espectroscopía infrarroja, Raman y de neutrones, entre otros métodos31.
Las simulaciones de MD se pueden utilizar para estudiar sistemas en varios niveles de resolución32,33,34. Los sistemas que tienen como objetivo caracterizar interacciones biomoleculares específicas, enlaces de hidrógeno y otros detalles de alta resolución se estudian con simulaciones de todos los átomos (AA). Por el contrario, las simulaciones de grano grueso (CG) agrupan los átomos en grupos funcionales más grandes para reducir el costo computacional y examinarla dinámica a mayor escala. Entre estos dos se encuentran las simulaciones de átomos unidos (UA), donde los átomos de hidrógeno se combinan con sus respectivos átomos pesados para acelerar el cálculo33,35. Las simulaciones MD son una poderosa herramienta para la exploración de la dinámica de las membranas lipídicas y sus interacciones con otras moléculas y pueden servir para proporcionar mecanismos a nivel molecular para procesos de interés en la interfaz de la membrana. Además, las simulaciones de MD pueden servir para acotar objetivos experimentales y predecir las propiedades macromoleculares de un sistema determinado en función de las interacciones microscópicas.
En resumen, dado un conjunto de coordenadas iniciales, velocidades y un conjunto de condiciones como temperatura y presión constantes, las posiciones y velocidades de cada partícula se calculan a través de la integración numérica del potencial de interacción y la Ley del movimiento de Newton. Esto se repite de forma iterativa, generando así una trayectoria de simulación30. Estos cálculos se realizan con un motor MD; entre varios paquetes de código abierto, GROMACS36 es uno de los motores más utilizados y el que describimos aquí. También incluye herramientas para el análisis y la construcción de las coordenadas iniciales de los sistemas a simular37. Otros motores MD incluyen NAMD38; CHARMM39 y AMBER40, que el usuario puede seleccionar a su propia discreción en función del rendimiento computacional de un sistema determinado. Es fundamental visualizar las trayectorias durante la simulación, así como para el análisis e interpretación de los resultados. Hay una variedad de herramientas disponibles; aquí discutimos la dinámica molecular visual (VMD) que ofrece una amplia gama de características, incluida la visualización tridimensional (3-D) con métodos expansivos de dibujo y coloreado, la visualización de datos volumétricos, la construcción, preparación y análisis de trayectorias de sistemas de simulación MD y la creación de películas de trayectoria sin límites en el tamaño del sistema, si la memoria está disponible41,42,43.
La precisión de la dinámica predicha entre los componentes del sistema está directamente influenciada por el FF elegido para la propagación de la trayectoria. Los esfuerzos empíricos de parametrización de FF son llevados a cabo por pocos grupos de investigación. Los FF más establecidos y comunes para la distrofia muscular incluyen CHARMM39, AMBER 40, Martini44, OPLS45 y SIRAH46. El campo de fuerza47 del CHARMM36 aditivo de todos los átomos (C36) se usa ampliamente para AA MD de sistemas de membrana, ya que reproduce con precisión los datos estructurales experimentales. Fue desarrollado originalmente por la comunidad CHARMM y es compatible con múltiples motores MD como GROMACS y NAMD. A pesar de las mejoras en los FF comunes, existe un esfuerzo continuo para mejorar los conjuntos de parámetros para permitir predicciones que reproduzcan fielmente los observables experimentales, impulsados por el interés en sistemas particulares de estudio48,49.
Un reto a la hora de simular membranas lipídicas es determinar la longitud de la trayectoria de simulación. Esto depende en gran medida de las métricas a analizar y del proceso que se pretenda caracterizar. Típicamente, las mezclas lipídicas complejas requieren más tiempo para alcanzar el equilibrio, ya que más especies deben tener tiempo suficiente para difundirse en el plano de la membrana y alcanzar una organización lateral estable. Se dice que una simulación está en equilibrio cuando la propiedad de interés ha alcanzado una meseta y fluctúa alrededor de un valor constante. Es una práctica común obtener al menos 100-200 ns de trayectoria equilibrada para llevar a cabo un análisis estadístico adecuado sobre las propiedades e interacciones de interés. Es común ejecutar simulaciones solo de membrana entre 200 y 500 ns, dependiendo de la complejidad de la mezcla de lípidos y la pregunta de investigación. Las interacciones proteína-lípidos suelen requerir tiempos de simulación más largos, entre 500 y 2000 ns. Algunos enfoques para acelerar el muestreo y la dinámica observable con sistemas de membranas son: (i) el modelo mimético de membrana altamente móvil (HMMM), que reemplaza los carbonos finales de los lípidos en la membrana con solvente orgánico para acelerar el muestreo50; y ii) la repartición de la masa de hidrógeno (HMR), que combina una fracción de las masas de los átomos pesados dentro de un sistema con las de los átomos de hidrógeno para permitir el uso de un paso de tiempo de simulación más grande51.
En el siguiente protocolo se describe un enfoque fácil de usar para principiantes para crear, ejecutar y analizar modelos de membrana realistas mediante AA MD. Dada la naturaleza de las simulaciones de MD, se deben ejecutar múltiples trayectorias para tener en cuenta la reproducibilidad y el análisis estadístico adecuado de los resultados. Es una práctica actual ejecutar un mínimo de tres réplicas por sistema de interés. Una vez que se han seleccionado las especies lipídicas para el organismo y el proceso de interés, se describen y resumen en la Figura 2 los pasos básicos para construir, ejecutar y analizar una trayectoria de simulación de un sistema solo de membrana.
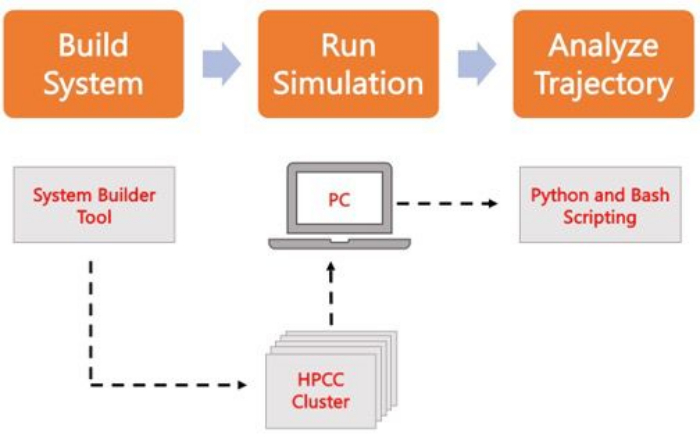
Figura 2. Esquema para ejecutar simulaciones de MD. Las casillas naranjas corresponden a los tres pasos principales descritos en el protocolo. Debajo está el flujo de trabajo del proceso de simulación. Durante la configuración del sistema, el sistema que contiene las coordenadas iniciales de un sistema de membrana solvatada se construye con un generador de entrada del sistema como CHARMM-GUI Membrane Builder. Después de transferir los archivos de entrada a un clúster informático de alto rendimiento, la trayectoria de simulación se propaga mediante un motor MD, como GROMACS. El análisis de la trayectoria se puede realizar en el clúster informático o en una estación de trabajo local junto con la visualización. A continuación, se lleva a cabo el análisis, ya sea utilizando paquetes con código de análisis incorporado, como GROMACS y VMD, o mediante scripts de Bash o varias bibliotecas de Python. Haga clic aquí para ver una versión más grande de esta figura.
