Lipitler, hücreler için sınırlar sağlayan ve hücre içi bölümlendirmeyisağlayan zarların ana bileşenleridir 1,2,3. Lipitler, bir polar baş grubu ve iki hidrofobik yağ asidi kuyruğu ile amfifiliktir; Bunlar, hidrofobik zincirlerin suile temasını en aza indirmek için çift tabaka halinde kendiliğinden birleşir 3,4. Hidrofilik baş gruplarının ve hidrofobik kuyrukların çeşitli kombinasyonları, biyolojik membranlarda gliserofosfolipidler, sfingolipidler ve steroller gibi farklı lipit sınıflarına neden olur (Şekil 1)1,5,6. Gliserofosfolipidler, gliserofosfat, uzun zincirli yağ asitleri ve düşük moleküler ağırlıklı baş gruplarındanoluşan ökaryotik hücre zarlarının birincil yapı taşlarıdır 7. Lipid isimlendirmesi, kafa gruplarındaki farklılıklara dayanır; örnekler arasında fosfatidil-kolin (PC), fosfatidil-etanolamin (PE), fosfatidil-serin (PS), fosfatidil-gliserol (PG), fosfatidil-inositol (PI) veya modifiye edilmemiş fosfatidik asit (PA)5,6. Hidrofobik kuyruklara gelince, omurga yapısı ile birlikte doygunluğun uzunluğu ve derecesi değişir. Olası kombinasyonlar çoktur ve memeli hücrelerinde binlerce lipit türü ile sonuçlanır6. Membran lipid bileşimindeki değişiklikler, hem integral membran proteinlerinin hem de periferik proteinlerinaktivitesini etkileyen farklı mekanik ve yapısal membran özelliklerine yol açar 2,6.
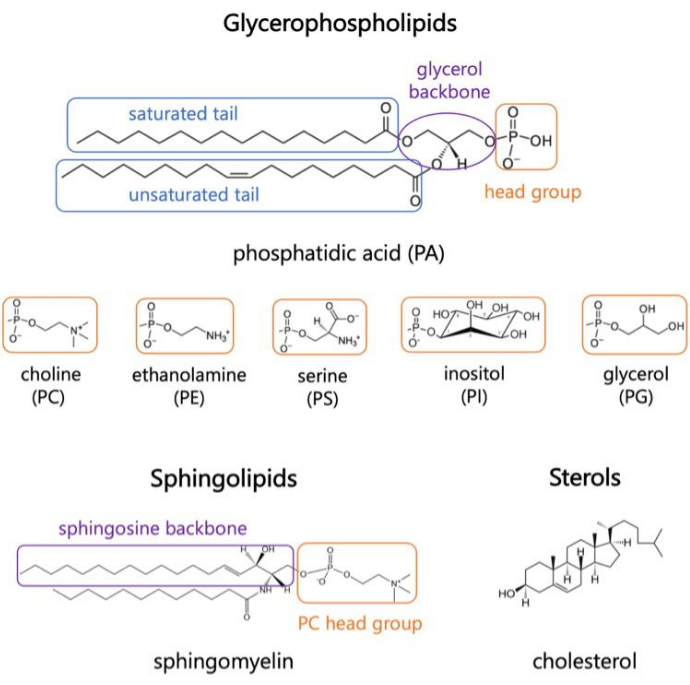
Şekil 1. Temsili lipit yapıları. Yağ asidi kuyrukları mavi kutularda, yaygın lipid baş grupları turuncu renkte ve örnek omurgalar mor renkte gösterilmiştir. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
Lipitler hücresel süreçlerde, sinyal kaskadlarında protein aktivasyonunda ve sağlıklı hücre homeostazında aktif oyunculardır 8,9. Değişen lipid dinamikleri enfeksiyonun sonucudur veyahastalığın patogenezinin belirteçleri olabilir 10,11,12,13,14,15. Hücre için engeller olarak, membran lipidlerinin incelenmesi ve bunların küçük moleküllerin nüfuz etmesindeki rolü, ilaç dağıtım sistemleri ve membran bozulma mekanizmalarıile ilgilidir 16,17. Organeller, dokular ve organizmalar arasındaki kimyasal çeşitlilik ve lipid türlerinin farklı oranları, karmaşık zar dinamiğine yol açar2. Bu nedenle, özellikle bir çalışmanın amacı diğer biyomoleküllerin zar ile etkileşimlerini incelemek olduğunda, lipid çift katmanlarının modelleme çalışmalarında bu özelliklerin korunması önemlidir. Bir modelde dikkate alınacak lipit türleri, ilgilenilen organizmaya ve hücresel bölmeye bağlıdır. Örneğin, PG lipidleri fotosentetik bateria18’de elektron transferi için önemlidir, fosforile inositol lipidleri (PIP’ler) ise plazma zarı (PM) dinamiklerinde ve memeli hücrelerinde sinyal kaskadlarında önemli oyunculardır 19,20. Hücrenin içinde PM, endoplazmik retikulum (ER), Golgi ve mitokondriyal zarlar, işlevlerini etkileyen benzersiz lipid bollukları içerir. Örneğin, ER, lipid biyogenezinin merkezidir ve kolesterolü PM ve Golgi’ye taşır; bol miktarda PC ve PE ile yüksek bir lipit çeşitliliği içerir, ancak membran akışkanlığınıdestekleyen düşük sterol içeriği 21,22,23,24 içerir. Buna karşılık, PM, organizmaya bağlı olarak yüzlerce ve hatta binlerce lipid türünü içerir25, hücre 24’teki diğer zarlara kıyasla karakteristik bir sertlik veren yüksek seviyelerde sfingolipidler ve kolesterol içerir. Yaprakçık asimetrisi, sfingomyelin, PC ve kolesterol açısından zengin bir dış yaprakçık ve basamakları24 sinyallemek için önemli olan PE, PI ve PS açısından zengin bir iç yaprakçık içeren PM gibi membranlar için dikkate alınmalıdır. Son olarak, lipid çeşitliliği, lipid salları olarak bilinen, paketleme ve iç düzende farklılık gösteren mikro alanların oluşumunu da tetikler24,26; Bunlar yanal asimetri sergiler, hücresel sinyalleşmede önemli roller oynadığı varsayılmaktadır26 ve geçici yapıları nedeniyle incelenmesi zordur.
Biyomoleküllerin membranlarla etkileşimlerini araştırmak için floroskopi, spektroskopi gibi deneysel teknikler ve dev unilameller veziküller (GUV’ler) gibi model membran sistemleri kullanılmıştır. Bununla birlikte, ilgili bileşenlerin karmaşık ve dinamik doğasını yalnızca deneysel yöntemlerle yakalamak zordur. Örneğin, proteinlerin transmembran alanlarının görüntülenmesi, bu tür çalışmalarda kullanılan membranların karmaşıklığı ve ilgilenilen işlem sırasında ara veya geçici durumların tanımlanması konusunda sınırlamalar vardır27,28,29. 1980’lerde lipid tek tabakalarının ve çift tabakalarının moleküler simülasyonunun ortaya çıkmasından buyana29, lipit-protein sistemleri ve etkileşimleri artık moleküler düzeyde ölçülebilir. Moleküler dinamik (MD) simülasyonu, moleküller arası kuvvetlerine dayalı olarak parçacıkların hareketini tahmin eden yaygın bir hesaplama tekniğidir. Bir katkı etkileşim potansiyeli, sisteminparçacıkları arasındaki bağlı ve bağlı olmayan etkileşimleri tanımlar 30. Bu etkileşimleri modellemek için kullanılan parametreler kümesi, simülasyon kuvvet alanı (FF) olarak adlandırılır. Bu parametreler, ab initio hesaplamalarından, yarı ampirik ve kuantum mekaniksel hesaplamalardan elde edilir ve diğer yöntemlerin yanı sıra X-ışını ve elektron kırınım deneyleri, NMR, kızılötesi, Raman ve nötron spektroskopisinden elde edilen çoğaltılan verilere optimize edilir31.
MD simülasyonları,32,33,34 çözünürlüğün çeşitli seviyelerindeki sistemleri incelemek için kullanılabilir. Spesifik biyomoleküler etkileşimleri, hidrojen bağlarını ve diğer yüksek çözünürlüklü ayrıntıları karakterize etmeyi amaçlayan sistemler, tüm atom (AA) simülasyonları ile incelenir. Buna karşılık, kaba taneli (CG) simülasyonlar, hesaplama maliyetini azaltmak ve daha büyük ölçekli dinamikleri incelemek için atomları daha büyük fonksiyonel gruplara ayırır33. Bu ikisi arasında, hesaplamayı hızlandırmak için hidrojen atomlarının ilgili ağır atomlarıyla birleştirildiği birleşik atom (UA) simülasyonlarıbulunur 33,35. MD simülasyonları, lipid membranlarının dinamiklerini ve bunların diğer moleküllerle etkileşimlerini araştırmak için güçlü bir araçtır ve membran arayüzünde ilgilenilen süreçler için moleküler düzeyde mekanizmalar sağlamaya hizmet edebilir. Ek olarak, MD simülasyonları, deneysel hedefleri daraltmaya ve mikroskobik etkileşimlere dayalı olarak belirli bir sistemin makromoleküler özelliklerini tahmin etmeye hizmet edebilir.
Kısaca, bir dizi başlangıç koordinatı, hız ve sabit sıcaklık ve basınç gibi bir dizi koşul verildiğinde, her parçacığın konumları ve hızları, etkileşim potansiyelinin ve Newton’un Hareket Yasasının sayısal entegrasyonu yoluyla hesaplanır. Bu, yinelemeli olarak tekrarlanır, böylece bir simülasyon yörüngesi30 oluşturulur. Bu hesaplamalar bir MD motoru ile gerçekleştirilir; Birkaç açık kaynaklı paket arasında, GROMACS36 en yaygın kullanılan motorlardan biridir ve burada açıkladığımız motordur. Ayrıca, simüle edilecek sistemlerin ilk koordinatlarının analizi ve oluşturulması için araçlar içerir37. Diğer MD motorları arasında NAMD38; CHARMM39 ve AMBER40, kullanıcının belirli bir sistemin hesaplama performansına dayalı olarak kendi takdirine bağlı olarak seçebileceği. Simülasyon sırasında yörüngeleri görselleştirmenin yanı sıra sonuçların analizi ve yorumlanması için de kritik öneme sahiptir. Çeşitli araçlar mevcuttur; burada, geniş çizim ve renklendirme yöntemleriyle üç boyutlu (3-D) görselleştirme, hacimsel veri görselleştirme, MD simülasyon sistemlerinin yörüngelerini oluşturma, hazırlama ve analiz etme ve bellek mevcutsa sistem boyutunda herhangi bir sınırlama olmaksızın yörünge filmi yapımı dahil olmak üzere çok çeşitli özellikler sunan görsel moleküler dinamikleri (VMD) tartışıyoruz41,42,43.
Sistem bileşenleri arasında tahmin edilen dinamiklerin doğruluğu, yörüngenin yayılması için seçilen FF’den doğrudan etkilenir. Ampirik FF parametrelendirme çabaları az sayıda araştırma grubu tarafından sürdürülmektedir. MD için en köklü ve yaygın FF CHARMM39, AMBER 40, Martini44, OPLS 45 ve SIRAH 46’dır. Tüm atom katkı maddesi CHARMM36 (C36) kuvvet alanı47, deneysel yapısal verileri doğru bir şekilde yeniden ürettiği için membran sistemlerinin AA MD’si için yaygın olarak kullanılmaktadır. Başlangıçta CHARMM topluluğu tarafından geliştirilmiştir ve GROMACS ve NAMD gibi birden fazla MD motoruyla uyumludur. Yaygın FF’lerdeki gelişmelere rağmen, belirli çalışma sistemlerine olan ilgiler tarafından yönlendirilen, deneysel gözlemlenebilirleri yakından yeniden üreten tahminlere izin vermek için parametre setlerini geliştirmek için sürekli bir çaba vardır48,49.
Lipid membranları simüle ederken karşılaşılan bir zorluk, simülasyon yörüngesinin uzunluğunu belirlemektir. Bu, büyük ölçüde analiz edilecek metriklere ve karakterize etmeyi amaçlayan sürece bağlıdır. Tipik olarak, karmaşık lipid karışımlarının dengeye ulaşması için daha uzun zaman gerekir, çünkü daha fazla türün zar düzleminde yayılması ve kararlı bir yanal organizasyona ulaşması için yeterli zamana sahip olması gerekir. İlgilenilen özellik bir platoya ulaştığında ve sabit bir değer etrafında dalgalandığında bir simülasyonun dengede olduğu söylenir. İlgilenilen özellikler ve etkileşimler üzerinde uygun istatistiksel analiz yapmak için en az 100-200 ns dengeli yörünge elde etmek yaygın bir uygulamadır. Lipid karışımının karmaşıklığına ve araştırma sorusuna bağlı olarak 200-500 ns arasında yalnızca membran simülasyonları çalıştırmak yaygındır. Protein-lipid etkileşimleri tipik olarak 500-2000 ns arasında daha uzun simülasyon süreleri gerektirir. Membran sistemleriyle örneklemeyi ve gözlemlenebilir dinamikleri hızlandırmak için bazı yaklaşımlar şunlardır: (i) örneklemeyi hızlandırmak için membrandaki lipitlerin uç karbonlarını organik çözücü ile değiştiren oldukça hareketli membran mimetik (HMMM) modeli50; ve (ii) daha büyük bir simülasyon zaman adımı51’in kullanılmasına izin vermek için bir sistem içindeki ağır atomların kütlelerinin bir kısmını hidrojen atomlarınınkilerle birleştiren hidrojen kütle yeniden bölümleme (HMR).
Aşağıdaki protokol, AA MD kullanarak gerçekçi membran modelleri oluşturmak, çalıştırmak ve analiz etmek için yeni başlayanlar için uygun bir yaklaşımı ele almaktadır. MD simülasyonlarının doğası göz önüne alındığında, tekrarlanabilirliği ve sonuçların uygun istatistiksel analizini hesaba katmak için birden fazla yörünge çalıştırılmalıdır. İlgilenilen sistem başına en az üç çoğaltma çalıştırmak geçerli bir uygulamadır. İlgilenilen organizma ve süreç için lipit türleri seçildikten sonra, yalnızca zardan oluşan bir sistemin simülasyon yörüngesini oluşturmak, çalıştırmak ve analiz etmek için temel adımlar Şekil 2’de özetlenir ve özetlenir.
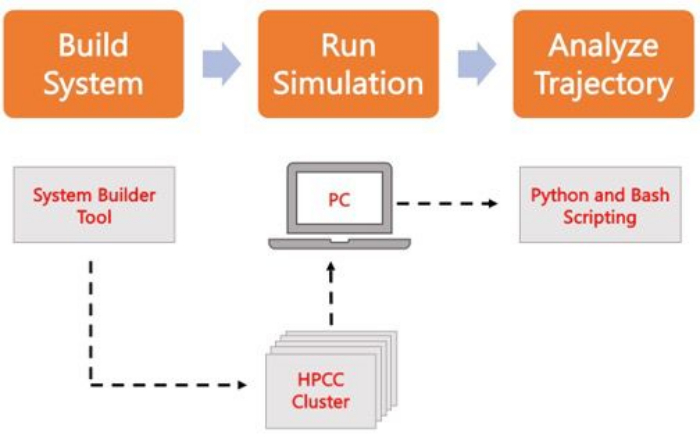
Şekil 2. MD simülasyonlarını çalıştırmak için şematik. Turuncu kutular, protokolde açıklanan üç ana adıma karşılık gelir. Aşağıda simülasyon sürecinin iş akışı yer almaktadır. Sistem kurulumu sırasında, çözülmüş bir membran sisteminin başlangıç koordinatlarını içeren sistem, CHARMM-GUI Membrane Builder gibi bir sistem giriş üreteci ile oluşturulur. Giriş dosyalarını yüksek performanslı bir bilgi işlem kümesine aktardıktan sonra, simülasyon yörüngesi GROMACS gibi bir MD motoru kullanılarak yayılır. Yörünge analizi, görselleştirme ile birlikte bilgisayar kümesinde veya yerel bir çalışma istasyonunda yapılabilir. Analiz daha sonra GROMACS ve VMD gibi yerleşik analiz koduna sahip paketler kullanılarak veya Bash komut dosyaları veya çeşitli Python kitaplıkları kullanılarak gerçekleştirilir. Bu rakamın daha büyük bir sürümünü görüntülemek için lütfen buraya tıklayın.
