The methodology designed in this paper is specifically intended to enhance the preparation of embryo samples for single-cell omics from E6.5 to E8. This robust pipeline consists of five major steps: synchronized timed pregnancies, embryo isolations, same-day genotyping, cell dissociation, and assessment of cell viability (Figure 1A). While the presented data focuses on time points from E7 to E7.5, it can be applied to embryos up to E8 (Figure 1B) with small variations in the procedure (referred to notes throughout the protocol). Synchronized timed pregnancies were achieved by placing two female mice into a cage with a male mouse. Vaginal plugs were checked every morning before 10 AM, and if a plug was observed, it was considered E0.5 by noon of that day (Figure 2A). In this study, optimal conditions for plugs required a female mouse in the estrus or proestrus stage to be paired with a male with a history of successfully placing several plugs during prior breeding (Figure 2B). Only obvious plugs were considered for embryo isolations (Figure 2C).
The developmental timing was estimated following the timetable in Figure 2A. For E7.5, embryo dissection started at 12 PM on the 7th day following the day of the detection of a plug. Figure 3 exemplifies a successful embryo isolation at E7.5. After the pregnant dam was euthanized, the uterine horn was dissected, the decidual swelling was individually cut, revealing the embryo, and the yolk sack was isolated for genotyping (Figure 3).
Same-day genotyping was performed within 3 h of embryo isolation following the steps in Figure 4A. The embryos were kept on ice during the process of genotyping to preserve their integrity. Do not freeze the embryos, as it will decrease the number of viable cells per embryo. Figure 4B,C show the visceral yolk sack, the parietal endodermal sac, and the ectoplacental cone with associated maternal blood. The visceral yolk sac was utilized for genotyping (Figure 4C). After digesting the yolk sac, the PCR mix was prepared, and the PCR reaction was run. The resulting PCR product was then separated on an agarose gel. Figure 4D shows expected fragment sizes for the LoxP and wild-type alleles (597 bp and 498 bp, respectively) and the Cre allele (650 bp). In Figure 4E, an example of yolk sac genotyping from 6 embryos obtained from breeding pairs carrying Cre and LoxP (flox) alleles is presented. The gels depict the PCR products of the Cre and LoxP alleles in the 6 embryos analyzed. Embryos number 2 and number 5 carry 2 flox alleles and 1 cre allele; therefore, they are considered conditional KO embryos (Figure 4E, red dots). Embryos 1 and 3 have 2 flox alleles but are negative for Cre; therefore, they are considered flox controls. Embryo 4 has 2 flox alleles and a faint band for the Cre allele, resulting in an "unclear" genotype. This embryo was not further processed (alternatively, the experimentalist may consider repeating the genotyping or conducting a secondary validation of the conditional KO using cells expressing Cre). It is important to note that the Cre driver used in this experiment is not expressed in the visceral yolk sac15; hence, the LoxP alleles do not appear shifted on the gel of Cre-positive embryos compared to the Cre-negative ones.
Cell count and good viability are required for a successful single-cell experiment. Suspensions with low cell viability, a high percentage of dead cells, clumping, or significant debris are unsuitable for further processing. Optimal conditions are for 700-1200 live cells per microliter and >90% viability. Figure 5A presents a panel illustrating both good and sub-optimal cell viability. The same criteria can also be applied for nuclei isolation, as shown in Figure 5B. However, it is crucial to note that the evaluation of trypan blue differs from cells and nuclei: viable cells do not incorporate trypan blue, while viable nuclei do. If cells/nuclei suspension is optimal (>90% viability), proceed with single-cell partitioning using a microfluidic chip following manufacturer's procedures11. Troubleshooting options are provided for suspensions where cell/nuclei viability is between 60%-89%, as depicted in Figure 5C. If viability, regardless of the total number of cells, falls below 60%, consider halting the experiment.
The entire pipeline from the culling of the pregnant dam to library construction takes a total of 8 hours on the same day (i.e., protocol steps 2 to 6 must be performed on the same day). Following the procedures outlined by the single-cell manufacturers' procedures11, library constructions were prepared for single-cell RNA sequencing using cells obtained from E7 embryos, with cell viability ranging from sub-optimal to optimal conditions, aiming for an estimated target recovery of 2000 cells (Figure 6). Figure 6A depicts a representative fragment size distribution of scRNAseq libraries for both sub-optimal and optimal conditions, indicating that cell viability does not significantly affect the entire single-cell partitioning process. The fragment size distribution ranged between 400-500 bp. This indicates that sub-optimal conditions do not affect the process of library preparation. Figure 6B shows the outcomes of both successful and sub-optimal single-cell RNAseq experiments. Following sequencing, quality control checks were conducted on samples and observed that, in cases where cell viability was sub-optimal, only 10% of cells were successfully sequenced. In contrast, optimal samples exhibited a higher percentage, with 91% of the total cells being sequenced. This is further proven by barcode plots, indicating that the sub-optimal conditions have larger background noise compared to optimal conditions. Clustering analysis was performed for both experiments and revealed 9 clusters in the optimal conditions and 4 in the sub-optimal. Annotation of the clusters using known markers3 revealed expected cell -types in the E7 embryos, including epiblast and primitive streak (Figure 6B). Cluster annotations in the suboptimal experiment were not possible due to the lack of enrichment in known markers for each cluster. This highlights the importance of high-quality cells required for the proper representation of data during these stages of development.
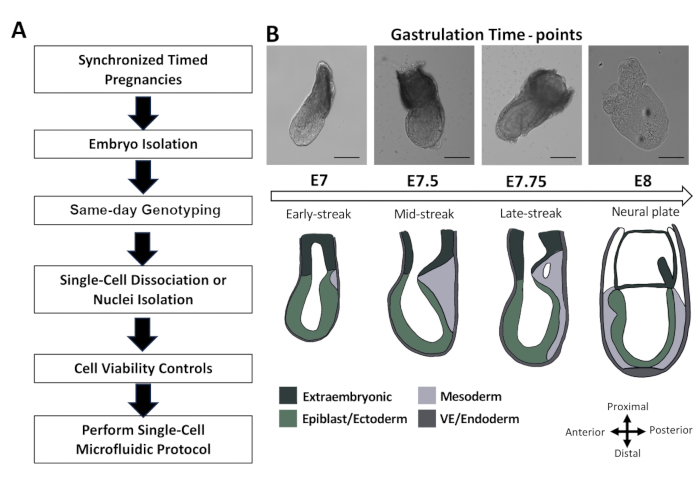
Figure 1: Optimization of gastrulating whole mouse embryos for single-cell RNA sequencing. (A) Workflow schematic for obtaining high-quality cells and/or nuclei from gastrulating embryos. (B) Representative bright field images and adapted scheme3 of mouse embryos during gastrulation from E7 to E8. Scale bar: 125 µm. Please click here to view a larger version of this figure.
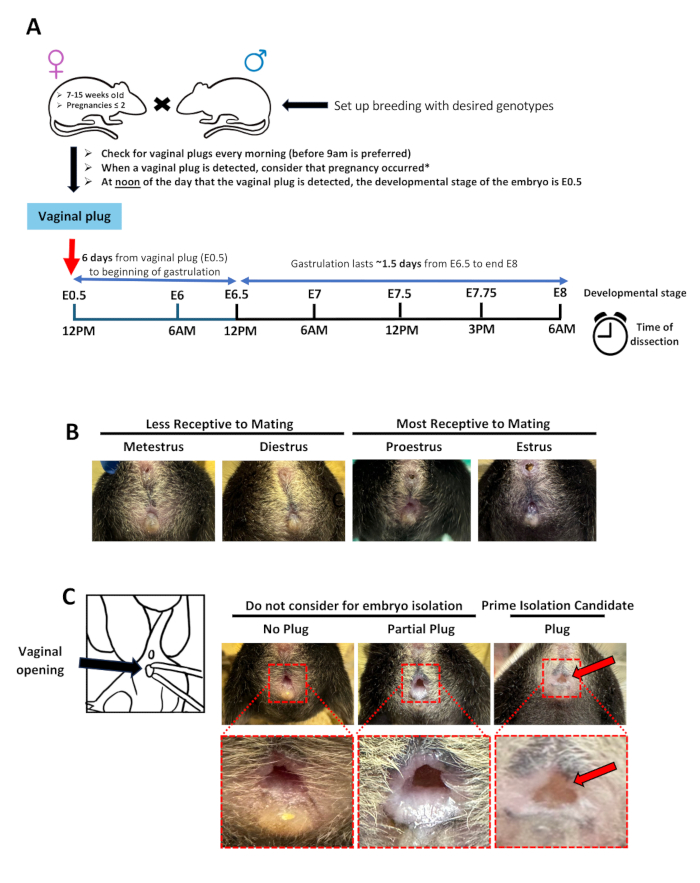
Figure 2: Strategy for efficient synchronized timed pregnancies for embryo isolations during gastrulation. (A) Schematic diagram indicating the pipeline for timed pregnancies and a timetable describing the times from plug detection to embryo isolation for analysis of specific gastrulation phases. (B) Representative images of the phases of the estrus cycle for female mice. The phases most receptive to breeding are indicated. (C) Schematic diagram illustrating how to check for vaginal plugs and examples of vaginal openings without a plug, a partial plug, or a good plug to consider for embryo isolations. The vaginal openings are zoomed in below each image. The plug (coagulated semen in the vagina opening) is highlighted with a red arrow pointing to it. Please click here to view a larger version of this figure.
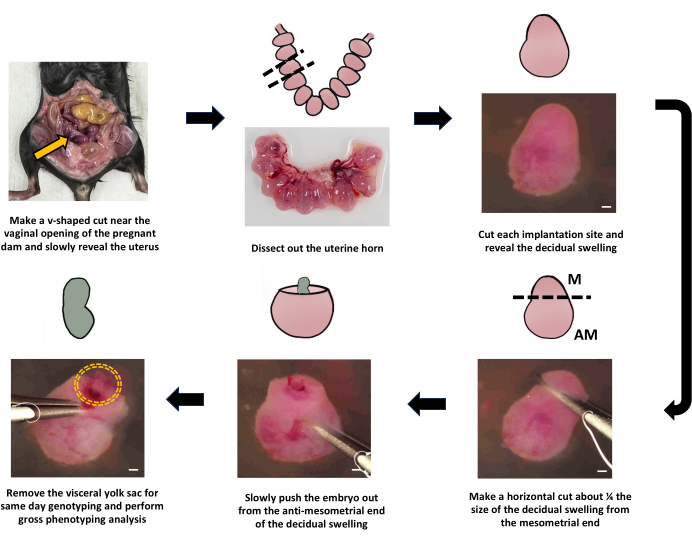
Figure 3: Dissection and genotyping of E7.5 embryos for single-cell RNA sequencing. Schematic diagrams and images of the process of isolating E7.5 embryos and dissecting the visceral yolk sac. The yellow arrow indicates the uterus of the pregnant dam, and the yellow dashed circle outlines the location of the embryo. Dashed lines represent the areas that were cut during dissection. "M" denotes the mesometrial end, while "AM" denotes the anti-mesometrial end. Scale bars in stereoscope images are 400 µm. Please click here to view a larger version of this figure.
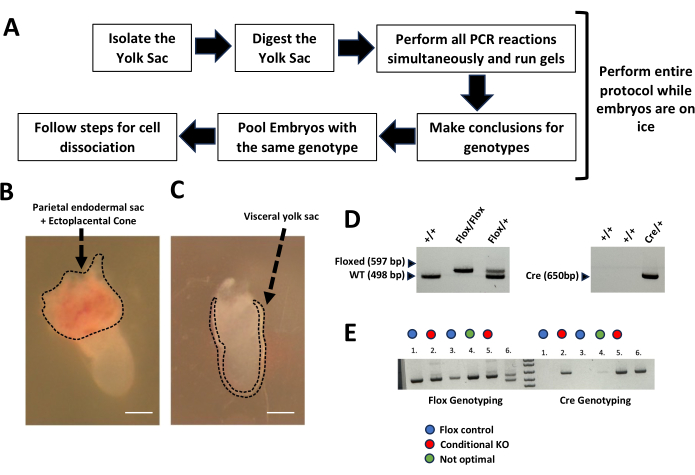
Figure 4: Same-day genotyping of yolk sacs from gastrulating mouse embryos. (A) Workflow schematic for same-day genotyping of yolk sacs. (B) Representative image of the parietal endodermal sac and ectoplacental cone with associated maternal blood. (C) Visceral yolk sac in E7.5 embryos highlighted by the dashed line. Scale bar: 200 µm. (D) Representative gels of wild type (+/+), heterozygous (Flox/+), and homozygous flox (Flox/Flox), and Cre genotyping (Mesp1cre15) with observed DNA fragment sizes. (E) Same-day genotyping results for Cre and flox alleles from yolk sacs of 6 embryos (1-6) are provided. The two embryos with flox/flox and Cre negative (-) genotyping are flox controls (blue dots), while those with flox/flox and Cre positive (+) genotyping are conditional KOs (red dots). Embryo 4 is not optimal due to the faint Cre band (flox/flox and Cre-unclear) and is consequently excluded from further processing. Embryos with the same genotype are pooled and processed for single-cell RNA sequencing. Note that the presence of the Cre allele in the yolk sac does not affect the size of the flox bands , as the Cre used (Mesp1cre) is not expressed in the yolk sac. Please click here to view a larger version of this figure.
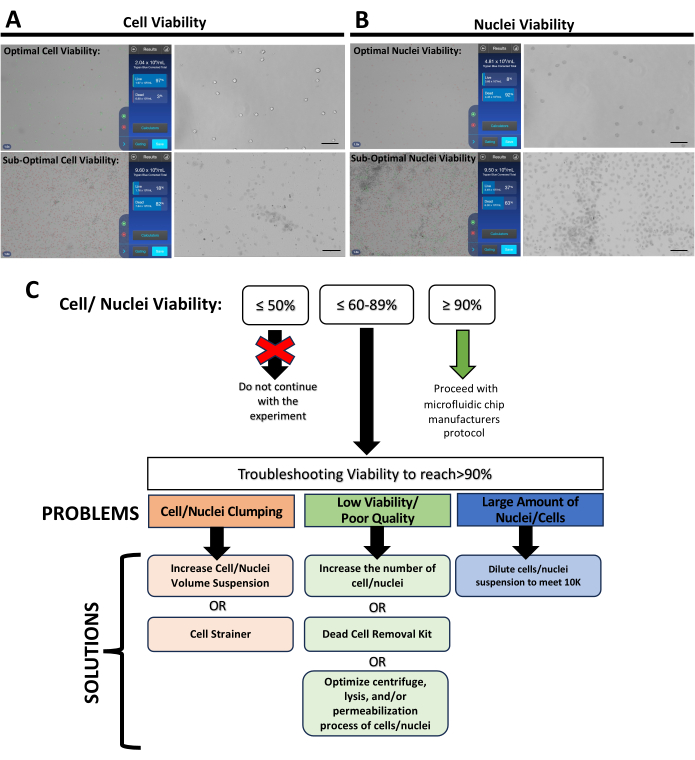
Figure 5: Assessment of cell quality and nuclei viability. (A) Representative images of optimal and sub-optimal cell viability conditions from E7.5 embryos. (B) Representative images of optimal and sub-optimal nuclei viability conditions from E8 embryos (C) Troubleshooting scheme indicating potential solutions to help increase the viability of cells before starting single-cell partitioning. Please click here to view a larger version of this figure.
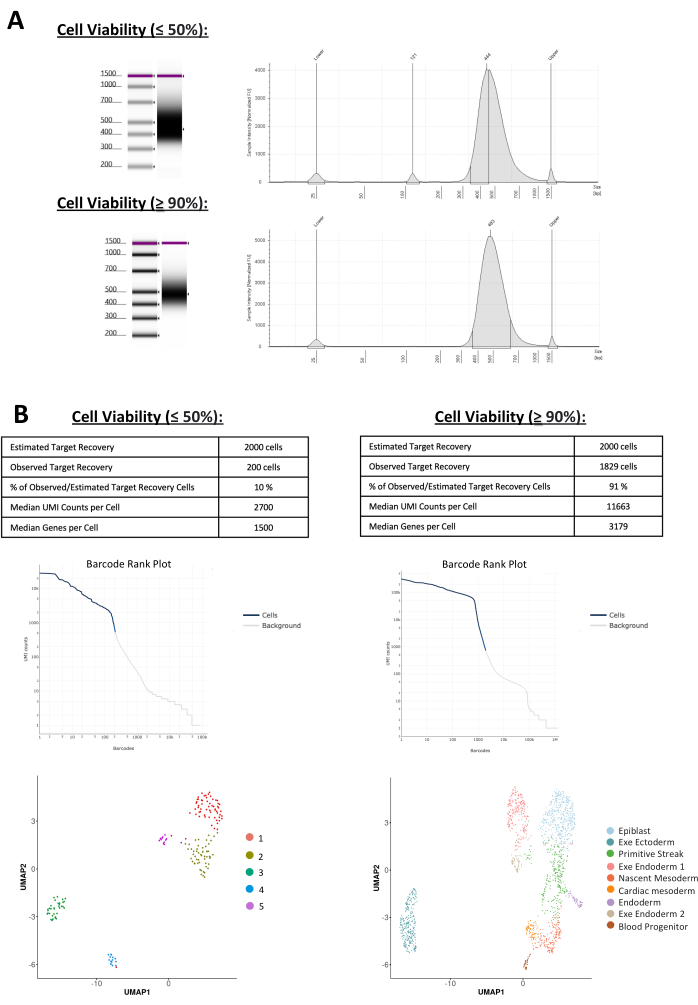
Figure 6: Single-cell RNA sequencing of E7 mouse embryos. (A) Representative trace of fragment size distribution for single-cell RNA sequencing libraries from E7 mouse embryos for both sub-optimal and optimal conditions of cell viability, with the main peak near 400-500 bp. Note that the traces are similar between these conditions, indicating that the cell viability does not affect the quality controls of the library. (B) Analysis of single-cell RNA sequencing outcomes for E7 mouse embryos under both sub-optimal and optimal conditions showing observed target recovery, barcode ranking, and clustering of cell types through uniform manifold approximation and projection (UMAP) distribution. Please click here to view a larger version of this figure.
| Lysis Buffer | |||
| Reagent Name | Stock Concentration | Final Concentration | Total Volume 1 mL |
| Tris-HCL (Ph7) | 1 M | 10 mM | 10 µL |
| NaCl | 5 M | 10 mM | 2 µL |
| MgCl2 | 0.1 M | 3 mM | 3 µL |
| Tween-20 | 20% | 0.10% | 5 µL |
| Nondiedt P40 | 10% | 0.10% | 10 µL |
| Digitonin | 5% | 0.01% | 2 µL |
| DTT | 1 M | 1 mM | 1 µL |
| RNase inhibitor | 40 U/µL | 1 U/µL | 25 µL |
| Nuclease-free Water | — | — | 924 µL |
| Wash Buffer | |||
| Reagent Name | Stock Concentration | Final Concentration | Total Volume 1 mL |
| BSA in PBS | 10% | 1% | 100 µL |
| PBS | — | — | 900 µL |
Table 1: Nuclei isolation lysis buffer and wash buffer composition.
