Source : Michael S. Lee1 et Tonya J. Webb1
1 Département de microbiologie et d’immunologie, University of Maryland School of Medicine et Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
L’immunohistochimie (IHC) et l’immunocytochimie (ICC) sont des techniques utilisées pour visualiser l’expression et la localisation d’antigènes spécifiques à l’aide d’anticorps. La première utilisation publiée de l’IHC a été en 1941 quand Albert Coons a utilisé la technique pour visualiser la présence d’antigène pneumococcique dans les sections de tissus de souris infectées par Pneumococcus (1). Le nom, immunohistochimie, est dérivé des racines “immuno-,” en référence aux anticorps, et “histo-,” en référence aux sections de tissu utilisées dans IHC. La racine “cyto-” dans l’immunocytochimie met en évidence la différence clé entre l’ICC et IHC. Alors que le CiSE utilise des sections de tissus entiers, l’ICC utilise des cellules qui ont été isolées des tissus ou cultivées en culture. La différence dans les échantillons utilisés signifie que la préparation des échantillons diffère techniquement entre le CSI et l’ICC, mais sinon les protocoles de l’ICC et du CiSE sont identiques et on constatera que les termes sont fréquemment utilisés de manière interchangeable.
Au CSI et à l’ICC, des anticorps munis d’étiquettes chimiques ou fluorescentes, comme la peroxidase ou la rhodamine, respectivement, sont utilisés pour visualiser la distribution de tout antigène d’intérêt par une liaison spécifique de l’anticorps étiqueté à l’antigène. Dans le cas du CSI, de fines tranches de tissu sont immobilisées sur une glissière pour maintenir la structure du tissu avant d’être tachées, ce qui permet la visualisation d’antigènes dans le contexte de tissus entiers (figure 1). Dans le cas de l’ICC, les cellules sont réparties uniformément sur une diapositive avant d’être tachées, ce qui permet la visualisation de la distribution d’antigènes dans les cellules individuelles, mais pas dans la structure d’un tissu spécifique. En raison des similitudes entre les deux protocoles, ce protocole mettra l’accent sur le CSI afin d’aborder les complexités additionnelles de la préparation des échantillons impliquées dans le CSI.
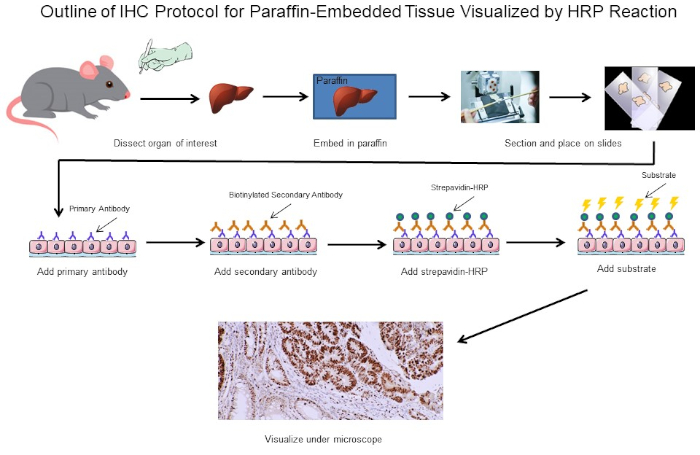
Figure 1 : Aperçu du Protocole du CSI. Contour visuel d’un protocole d’IHC pour le tissu paraffine-incorporé disséqué d’une souris. Ce protocole utilise un anticorps secondaire biotinylated et la strepavidtine-HRP pour visualiser l’emplacement de la liaison d’anticorps. D’autres options, telles que des anticorps étiquetés fluorescents, sont également possibles. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
La première décision importante lors de l’exécution du CSI est de savoir comment préparer les sections tissulaires afin de maintenir la structure du tissu tout au long du processus de coloration. Les deux principaux choix sont des sections fixées par formaline de tissu incorporé à la paraffine ou des sections fraîches de tissu congelé. Il n’y a pas de réponse simple quant à la méthode à utiliser car elle dépend de l’analyse en aval qui sera effectuée. On pense généralement que la fixation formaline des tissus incorporés à la paraffine est généralement mieux préservée de la morphologie tissulaire pour une imagerie optimale, tandis que la congélation des tissus frais peut préserver la fonction protéique pour les essais ultérieurs à l’extérieur du CSI. En outre, il a été démontré que les sections de tissus congelés frais conviennent mieux à l’analyse de l’expression génique (2). Une troisième considération est de savoir si oui ou non les anticorps pour votre antigène d’intérêt sont adaptés pour les sections de tissus fixes ou congelés, comme certains anticorps ont seulement été optimisés pour un type spécifique de section et peut ne pas fonctionner pour d’autres. Enfin, il faut aussi déterminer combien de temps ils ont besoin pour stocker les sections de tissus, car les échantillons congelés frais doivent être conservés à -80 oC et ne peuvent pas durer au-delà d’un an tandis que les sections fixes peuvent être stockées beaucoup plus longtemps à température ambiante. Ce sont quelques-unes des principales considérations pour déterminer s’il faut utiliser des sections fixées par formaline de tissu incorporé à la paraffine ou des sections fraîches de tissu congelé. En fin de compte, si l’on a assez de tissu, il peut être préférable juste d’avoir quelques-uns des deux.
Dans cette expérience, nous avons entrepris de déterminer si l’expression de cyclin D1 a été augmentée dans les rates agrandies d’un modèle spontané de souris du développement de lymphome. Des échantillons de tissu splénique ont d’abord été isolés de souris de type sauvage, de souris transgéniques qui n’ont pas de lymphome, ou de souris transgéniques qui ont développé spontanément un lymphome. Les échantillons de tissu de rate ont été fixés dans le paraformaldéhyde, incorporés dans le paraffine, sectionné, souillé utilisant un anticorps primaire anti-cyclin d’anticyclin de souris suivi d’un anticorps secondaire d’anti-souris de cheval, et développé utilisant 3,3-diaminobenzidine (DAB). Les sections ont ensuite été contre-tachées dans Harris Hematoxylin Solution, puis les sections ont été imaged à 20X grossissement.
Réactifs
Sections paraffines
- 4% Paraformaldéhyde (PFA)
- Éthanol (anhydrous dénaturé, grade histologique 100%, 95%, 80%, 75% et 50%). Peut être dilué à partir de 100% de stock à l’aide d’eau double distillée (ddH2O)
- Xylène
- Lamlede de verre compatible iHC pour s’assurer que la section tissulaire reste attachée tout au long de la procédure. Les lames de verre compatibles IHC ont un revêtement spécialisé et sont facilement disponibles auprès de plusieurs détaillants. Si vous effectuez icc, utilisez une diapositive chambrée. Les glissières chambrées permettent d’ensemencées dans les chambres et placées dans l’incubateur jusqu’à ce que les cellules se fixent à la glissière et atteignent la confluence appropriée, à ce moment-là les chambres peuvent être enlevées et la coloration peut se dérouler de la même manière que le CSI.
- pétrole
- 0,3 % Peroxyde d’hydrogène (H2O2)/méthanol : Pour préparer, ajouter 1 ml 30 % H2O2 à 99 ml de méthanol. Magasinez à -20oC
- Tampon de récupération d’antigène : IHC citrate buffer pH 6.0
Sections fraîches congelées
- Composé optimal de température de coupe (OCT)
- Fixation optimale : 4 % de PFA ou d’acétone refroidi à -20 oC
Coloration
- Tampon de blocage : doit être déterminé par l’utilisateur. Un exemple est le sérum de cheval dilué dans 1X PBS
- Anticorps primaires dilués : voir les spécifications du fabricant
- Anticorps secondaires biotinylated dilués : voir les spécifications du fabricant
- Peroxidase avide-raifort dilué (HRP) : Seulement pour la visualisation de peroxidase. Voir les spécifications du fabricant.
- DAB ou un autre substrat compatible
- Counterstain (facultatif)
- Éthanol (anhydre dénaturée, grade histologique 100% et 95%)
- Xylène
- Mont Organo/Limonène
IHC and ICC have a vast range of applications. For example, one use of IHC is to examine the expression of oncogenes in spontaneous mouse models of tumor development. In Figure 2, we set out to determine if cyclin D1 expression was increased in enlarged spleens in a spontaneous mouse model of lymphoma development. Splenic tissue samples were fixed in paraformaldehyde, embedded in paraffin, sectioned, stained using an anti-cyclin D1 antibody (diluted 1:200 in blocking buffer), and then the sections were imaged at 20X magnification. Cyclin D1 expressing cells are indicated by the reddish-brown color against the blue tissue background. These results suggest that cyclin D1 expression was increased in enlarged spleens, indicating a correlation between cancer development and cyclin D1 expression in this model.
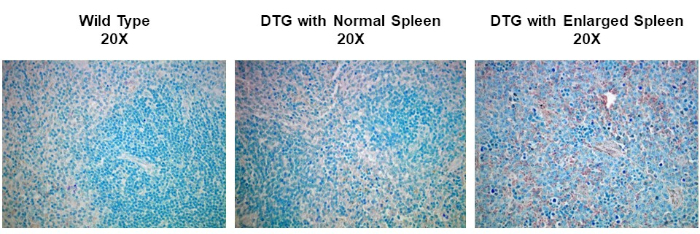
Figure 2: Splenic Cyclin D1 Expression in a Spontaneous Double Transgenic (DTG) Mouse Model of Lymphoma. An image of splenic tissue stained with an anti-Cyclin D1 primary antibody, counterstained with methyl green, and visualized using a biotinylated secondary antibody and ABC reagent activated with DAB substrate. The reddish-brown color represents locations where the antibody has bound indicating the presence of Cyclin D1 expressing tumor cells within the structure of splenic tissue that has been counterstained blue. Please click here to view a larger version of this figure.