מקור: דומיניק ר. בולינו1, אריק א. לגנצוב2, טוניה ג’יי ווב1
1 המחלקה למיקרוביולוגיה ואימונולוגיה, בית הספר לרפואה של אוניברסיטת מרילנד ומרכז הסרטן המקיף מרלן וסטיוארט גרינבאום, בולטימור, מרילנד 21201
2 המרכז להנדסה וטכנולוגיה ביו-רפואית, בית הספר לרפואה של אוניברסיטת מרילנד, בולטימור, מרילנד 21201
מיקרוסקופיית פלואורסצנטיות קונפוקלית היא טכניקת הדמיה המאפשרת רזולוציה אופטית מוגברת בהשוואה למיקרוסקופיה אפיפלואורסצנטית קונבנציונלית “רחבת שדה”. מיקרוסקופים קונפוקליים מסוגלים להשיג רזולוציה אופטית משופרת x-y באמצעות “סריקת לייזר” – בדרך כלל קבוצה של מראות מבוקרות מתח (מראות גלוונומטר או “גלבו”) המכוונות את תאורת הלייזר לכל פיקסל של הדגימה בכל פעם. חשוב מכך, מיקרוסקופים קונפוקליים משיגים רזולוציה z-צירית מעולה באמצעות חור סיכה כדי להסיר מאורת מיקוד שמקורה במיקומים שאינם במישור z נסרקים, ובכך מאפשרים לגלאי לאסוף נתונים ממישור z שצוין. בגלל רזולוציית z גבוהה בר השגה במיקרוסקופיה קונפוקלית, ניתן לאסוף תמונות מסדרה של z-planes (המכונה גם z-stack) ולבנות תמונה תלת-ממדית באמצעות תוכנה.
לפני דיון במנגנון של מיקרוסקופ קונפוקלי, חשוב לשקול כיצד מדגם אינטראקציה עם אור. האור מורכב מפוטונים, חבילות של אנרגיה אלקטרומגנטית. פוטון הפוחת במדגם ביולוגי יכול לקיים אינטראקציה עם המולקולות המרכיבות את המדגם באחת מארבע דרכים: 1) הפוטן אינו מתקשר ועובר דרך המדגם; 2) הפוטן משתקף / מפוזר; 3) הפוטון נספג על ידי מולקולה והאנרגיה הנספגת משתחררת כחום באמצעות תהליכים הידועים בשם ריקבון לא-רעיקטיבי; ו-4) הפוטן נספג והאנרגיה נספגת במהירות כפוטון משני בתהליך המכונה פלואורסצנטיות. מולקולה שהמבנה שלה מאפשר פליטת פלואורסצנטיות נקראת פלואורופור. רוב הדגימות הביולוגיות מכילות פלואורופורים אנדוגני זניחים; לכן פלואורופורים אקסוגניים חייבים לשמש כדי להדגיש תכונות של עניין במדגם. במהלך מיקרוסקופיית פלואורסצנטיות, המדגם מואר באור של אורך הגל המתאים לספיגה על ידי הפלואורופור. עם ספיגת פוטון, פלואורופור הוא אמר להיות “נרגש” ואת תהליך הקליטה מכונה “עירור”. כאשר פלואורופור מוותר על אנרגיה בצורה של פוטון, התהליך מכונה “פליטה”, והפוטון הנפלט נקרא פלואורסצנטיות.
קרן האור המשמשת לרגש פלואורופור מתמקדת בעדשה האובייקטיבית של מיקרוסקופ ומתכנסת ב”נקודת מוקד “שבה היא ממוקדת באופן מקסימלי. מעבר לנקודת המוקד האור שוב מתפצל. ניתן לדמיין את הקורות הנכנסות והיוצאות כצמד קונוסים הנגעים בנקודת המוקד (ראו איור 1, החלונית השמאלית). תופעת עקיפה מטילה מגבלה על מידת ההדוקה של קרן אור יכולה להיות ממוקדת – הקרן מתמקדת למעשה בנקודה בגודל סופי. שני גורמים קובעים את גודל נקודת המוקד: 1) אורך הגל של האור, ו -2) את יכולת איסוף האור של העדשה האובייקטיבית, המאופיינת בצמצם המספרי שלה (NA). המוקד “ספוט” משתרע לא רק במישור x-y, אלא גם בכיוון z, והוא למעשה נפח מוקד. הממדים של אמצעי אחסון מוקדי זה מגדירים את הרזולוציה המרבית הניתנת להשגה על-ידי הדמיה אופטית. למרות שמספר הפוטונים הוא הגדול ביותר בנפח המוקד, נתיבי האור החרוטיים מעל ומתחת למוקד מכילים גם צפיפות נמוכה יותר של פוטונים. כל פלואורופור בנתיב האור יכול אפוא להתרגש. במיקרוסקופיה אפיפלואורסצנטית קונבנציונלית (רחבת-שדה), פליטה מפלואורופורים מעל ומתחת למישור המוקד תורמת פלואורסצנטיות מחוץ לפוקוס (“רקע מעורפל”), מה שמפחית את רזולוציית התמונה ואת הניגודיות, כפי שמדגים באיור 1, כאשר הקובייה האדומה מייצגת את פליטת הפלורופור מעל המישור המוקדי (כוכב אדום) שגורמת לפלאורסצנטיות מחוץ לפוקוס (מימין למעלה). בעיה זו משופכת במיקרוסקופיה קונפוצלית, עקב ניצול של חור סיכה. (איור 2, מימין למטה). כפי שמתואר באיור 3, חור הסיכה מאפשר לפליטות שמקורן במוקד להגיע לגלאי (משמאל), תוך חסימת הפלואורסצנטיות מחוץ לפוקוס (מימין) מלהגיע לגלאי, ובכך לשפר הן את הרזולוציה והן את הניגודיות.
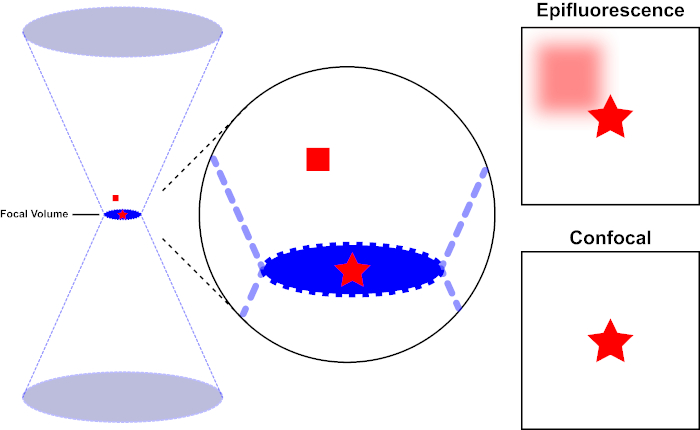
איור 1. רזולוציה אופטית של אפיפלואורסצנטיות לעומת מיקרוסקופיה קונפוקלית. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
קרן האור המשמשת לרגש פלואורופור מתמקדת בעדשה האובייקטיבית של מיקרוסקופ ומתכנסת בנפח מוקד ואז מתפצלת (משמאל). הכוכב האדום מייצג את מישור המוקד של דגימה שתמונה בעוד הריבוע האדום מייצג פליטת פלואורופור מעל מישור המוקד. בעת לכידת תמונה של מדגם זה באמצעות מיקרוסקופ אפיפלואורסצנטי, הפליטה מהכיכר האדומה מחוץ לפוקוס תהיה גלויה ותתרום ל”רקע מעורפל ” (מימין למעלה). למיקרוסקופים קונפוקליים יש חור סיכה המונע גילוי אור הנפלט מחוץ למישור המוקד, ומבטל את “הרקע המעורפל” (מימין למטה).

איור 2. אפקט חור הסיכה במיקרוסקופיה קונפוקלית. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
למרות שהעוצמה הגבוהה ביותר של אור העירור נמצאת במוקד העדשה (שמאל, אליפסה אדומה), חלקים אחרים של המדגם שאינם במוקד (ימין, כוכב אדום) יקבלו אור ופלואורסצ’ה. כדי למנוע אור הנפלט מאזורים אלה מחוץ לפוקוס כדי להגיע לגלאי, מסך עם חור סיכה נמצא מול הגלאי. רק האור בפוקוס (משמאל) הנפלט ממטוס המוקד מסוגל לנוע דרך חור הסיכה ולהגיע לגלאי. האור מחוץ לפוקוס (מימין) נחסם עם חור הסיכה ואינו מצליח להגיע לגלאי.
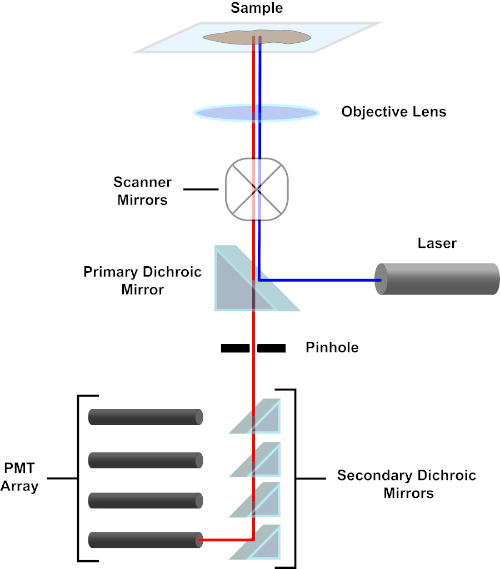
איור 3. רכיבים עיקריים של מיקרוסקופ סריקת לייזר קונפוקלי. אנא לחץ כאן כדי להציג גירסה גדולה יותר של איור זה.
למען הפשטות, התיאור המכניסטי של מיקרוסקופ קונפוקל יהיה מוגבל לזה של ניקון אקליפס Ti A1R. למרות ייתכנו הבדלים טכניים קלים בין מיקרוסקופים קונפוקליים שונים, A1R משמש היטב כמודל טוב לתיאור פונקציית מיקרוסקופ קונפוקלית. קרן אור העירור, המיוצרת על ידי מערך של לייזרי דיודה, משתקפת על ידי המראה הדיכרואית העיקרית אל המטרה, הממקדת את האור על הדגימה המצולמת. המראה הדיכרואית הראשית משקפת באופן סלקטיבי את אור העירור ומאפשרת לאור באורכי גל אחרים לעבור דרכם. לאחר מכן, האור נתקל במראות הסריקה שסוחפות את קרן האור על פני הדגימה באופן x-y, ומאירות פיקסל יחיד (x,Y) בכל פעם. פלואורסצנטיות הנפלטת על ידי פלואורופורים בפיקסל המואר נאספה על ידי העדשה האובייקטיבית ועוברת דרך המראה הדיכרואית הראשית כדי להגיע למערך של צינורות פוטומולטיפלייר (PMTs). מראות דיכרואיות משניות מכוונות את נורית הפליטה אל ה-PMT המתאים. אור העירור הפזור על ידי המדגם בחזרה למטרה משתקף על ידי המראה הדיכרואית הראשית בחזרה לכיוון הדגימה, ובכך נמנע מלהיכנס לנתיב אור הזיהוי ולהגיע ל-PMTs (ראו איור 3). זה מאפשר את הפלואורסצנטיות החלשה יחסית להיות כימות ללא זיהום על ידי אור מפוזרים מן קרן אור העירור, שהוא בדרך כלל סדרי גודל אינטנסיביים יותר מאשר פלואורסצנטיות. מכיוון שחור הסיכה חוסם אור מחוץ לנפח המוקד, האור המגיע לגלאי מגיע ממטוס zצר ונבחר. לכן, ניתן לאסוף תמונות מסדרה של מטוסי zסמוכים; סדרת תמונות זו מכונה לעתים קרובות ‘z-stack’. באמצעות התוכנה המתאימה, z-stack ניתן לעבד כדי ליצור תמונה 3D של הדגימה. יתרון מסוים של מיקרוסקופיה קונפוקלית הוא היכולת להבחין בין לוקליזציה תת-תאית של כתמים. לדוגמה, ההבחנה בין מכתים ממברנה מכתמים תאיים, וזה מאוד מאתגר עם מיקרוסקופיה אפיפלואורסצנטית קונבנציונלית (1, 2, 3).
הכנת מדגם היא היבט חשוב של הדמיה קונפוקלית. חוזק של טכניקות מיקרוסקופיה אופטית הוא הגמישות לתמונה של תאים חיים או קבועים. בעת ניסיון לייצר תמונות תלת-ממד, בגלל מספר התמונות שיש לרכוש עבור z-stack, הקושי לשמור על בריאות התא, ואת התנועה של תאים חיים organelles שלהם, השימוש בתאים קבועים אופייני. ההליך לתיקון והכתמת תאים לפלואורסצנטיות קונפוצלית דומה לזה המשמש באופן קונבנציונלי באימונופלואורסצנטיות. לאחר תרבית בשקופיות הקאמריות או על כיסויים, התאים קבועים באמצעות paraformaldehyde כדי לשמר את המורפולוגיה התאית. כריכת נוגדנים לא ספציפית נחסמת באמצעות אלבומין בסרום בקר, חלב או סרום רגיל. על מנת לשמור על הספציפיות של הנוגדנים המשניים, הפתרון המשמש לא צריך להיות מאותו מין שבו נוצרו הנוגדנים העיקריים. התאים דוגרים בנוגדנים ראשוניים שקושרים את האנטיגן של עניין. בעת סימון מספר מטרות תאיות, הנוגדנים העיקריים חייבים להיגזר כל אחד ממין אחר. נוגדנים המתייגים אנטיגן קשורים לאחר מכן בנוגדנים משניים מצומדים פלואורופור. נוגדנים משניים מצומדים פלואורופור יש לבחור כך שהם תואמים לאורכי הגל של עירור לייזר זמין במיקרוסקופ הקונפוקלי. כאשר מדמיינים אנטיגנים מרובים, ספקטרום העירור/ הפליטה של הפלורופורים צריך להיות שונה מספיק, כך האותות שלהם ניתן להפלות על ידי ניתוח מיקרוסקופי. הדגימה המוכתמת מותקנת לאחר מכן על שקופית להדמיה. מדיום הרכבה משמש למניעת התייבשות של פוטו-דגימות. אם תרצה, ניתן להשתמש במדיום הרכבה המכיל כתם נגד גרעיני (למשל DAPI או Hoechst) (4).
בפרוטוקול הבא, פיברובלסטים עכבר transfected כדי לבטא CD1d (LCD1) היו מוכתמים בנוגדנים המזהים CD1d ו CD107a (LAMP-1). CD1d הוא קומפלקס היסטו-תאימות גדול 1 (MHC 1)דמוי קולטן נוכח על פני השטח של אנטיגן מציג תאים המציגים אנטיגנים שומנים בדם. LAMP-1 (חלבון ממברנה הקשורים ליזומלית-1) הוא חלבון טרנס-מברן הנמצא בעיקר בממברנות ליזומליות. עבור מצגת אנטיגן נכונה, CD1d נסחר דרך תא ליסומלי pH נמוך, כך LAMP-1 משמש כסמן של התא הליסומלי עבור פרוטוקול זה. על ידי בדיקות תאי LCD1 עם אנטי CD1d ו- ANTI-LAMP-1 שיוצרו במינים שונים, נוגדנים משניים עם פלואורופורים ייחודיים יכולים לשמש כדי לקבוע את לוקליזציה של כל חלבון בתא והאם CD1d קיים בתאים ליזוזום חיובי LAMP-1.
In this experiment, mouse fibroblasts expressing the surface glycoprotein gene CD1d were fixed, immunostained and imaged on a confocal microscope. A representative image obtained using the above protocol is shown in Figure 4. In the top panel of A, single-channel images showing the staining pattern of each individual target are presented. These images comprise a single section (slice) of the z-stack captured. The right panel shows DAPI staining of nuclei of the cells. The center panels show CD1d stained in red and LAMP-1, a lysosomal marker, stained in green. The left panel is a composite image where the three different channels are merged. The appearance of yellow results from overlap of the red and green channels, and indicates an area where CD1d and LAMP-1 are co-localized. The results of the staining confirm that CD1d is localized in the LAMP-1+ endosomal compartments. There are also areas where only one color is present, which indicates the presence of CD1d or LAMP-1 without co-localization. The bottom panel of A shows a 3D rendering of the cells constructed from images captured in the z-stack.
Panel B shows a slice out of the z-stack at 100x magnification demonstrating the expression patterns of these two proteins in greater detail. The pink outlined box on the right side of the image displays the cross section of the x-coordinate designated by the pink line in the image, which represents the side view at the pink line. Similarly, the blue outlined box on the bottom of the image shows the cross section of the y-coordinate designated by the blue line in the image, which represents the front view at the blue line. The 3D rendering of the z-stack image enables users to view the image in 3D, visualizing all the x, y and z planes.
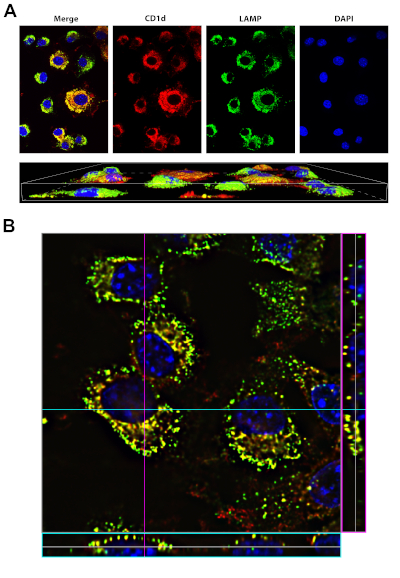
Figure 4: Staining of CD1d and LAMP1. Please click here to view a larger version of this figure.
A, top panel: LCD1 cells were fixed, permeabilized and stained with antibodies to CD1d (red) and LAMP-1 (green, a marker of the lysosomal compartment). DAPI (blue, was used to visualize the nucleus). The merge (left panel) shows that CD1d is localized in the LAMP-1 positive late endosomal/lysosomal compartment (yellow).
A, bottom panel: 3D rendering of the same cells in top panel. Images were acquired using a 40x oil-immersion objective on the Nikon Eclipse Ti, using the NIS Elements Advanced Research software.
B: 100x image of LCD1d cells stained as in A, with stack information for a particular y-coordinate (denoted by the blue line) on the bottom of the image (blue box). The stack information for a particular x-coordinate (denoted by the pink line) is shown on the right side of the image (pink box).
Confocal fluorescent staining is a relatively simple procedure that results in extremely high-quality images of specimens that are prepared in a similar way as for conventional fluorescence microscopy. In brief, samples are fixed, permeabilized, then blocked. Primary antibodies against a protein or proteins of interest are allowed to bind, then fluorophore-conjugated secondary antibodies are used to visualize the staining. Confocal fluorescence microscopy has applications in many areas of research. For example, by staining for markers of sub-cellular organelles along with a protein of interest, confocal microscopy can be used to determine the subcellular locations of diverse proteins. Compared to conventional fluorescence microscopy, confocal imaging can more effectively distinguish between cell surface and intracellular location of a protein. In addition, confocal imaging can also be used to determine whether two proteins co-localize within the cell. Although not outlined in this protocol, confocal fluorescence microscopy also can be performed on live cells to detect dynamic changes.
Video 1: Video created in NIS Elements Advanced Research software, highlighting the ability to move through the 3D rendering of the images. Please click here to view this video (Right click to download).