PEG precipitation increases EV yield
Our combination approach to EV isolation is significantly more efficient in terms of EV recovery as compared to traditional ultracentrifugation, as evident by the 90% reduction in the volume of starting material required. Ultracentrifugation, the current gold standard in EV isolation, requires approximately 100 mL of culture supernatant to produce an adequate EV prep for downstream assays, whereas our novel protocol requires only 10 mL. This reduction is made possible through the use of the PEG EV precipitation reagent which provides a significant increase in EV yield as measured by nanotracking analysis (NTA). The results in Figure 2A, which have been previously published6, indicate that when using 10 mL of culture supernatant PEG precipitation resulted in the recovery of 7.27 x 1010 EVs, which was approximately 500-fold more than the number of EVs recovered from the same starting material using ultracentrifugation (1.45 x 108). Increased efficiency of the isolation of exosomes, a specific EV subtype, was also observed as evident by increased levels of well-characterized exosome marker proteins, CD81, CD63, and CD9. Western blot analysis comparing EV preps produced from either ultracentrifugation or EV precipitation using a PEG precipitation reagent is shown in Figure 2B. These results show a 3,000-fold increase in CD81, a 4-fold increase in CD63, and a 40-fold increase in CD9 as measured by densitometry analysis. Taken together, these results suggest that the outlined protocol not only increases the total EV yield but also enhances recovery of exosomes when compared to ultracentrifugation.
Characterization of EVs and separation of EVs away from HIV-1 virus
In order to characterize the vesicles present in each fraction following nanoparticle enrichment, EVs were isolated from uninfected CEM or HIV-1 infected U1 cell culture supernatant using the outlined protocol and characterized using western blot of each nanoparticle-enriched iodixanol fraction. The data in Figure 3A show our previously published results which demonstrate the presence or absence of three exosomal tetraspanins (CD81, CD63, and CD9) in each fraction (CEM cells; top panel). The results indicate that exosomes, as defined by the presence of all three tetraspanins within the fraction, are found in three distinct populations: Exo #1 which includes fractions 10.8-12.0, Exo #2 which includes fractions 15.6-16.8, and Exo#3 which includes only the 18.0 fraction. Despite the presence of three distinct populations, the protocol cannot rule out the possibility of the presence of additional types of EVs within each fraction. Furthermore, these results do not definitively exclude the possibility that there are vesicles in these populations that are positive for a combination of the tested tetraspanin markers. These EVs could then be separated further by additional purification strategies.
The rapidly developing field of EV/exosome research has exploded since their discovery and has resulted in many newfound functions and applications for these vesicles. In particular, their role as intracellular messengers in not only normal physiology but also in disease has led to the use of significant resources to dissect the effects and utility of extracellular vesicles, specifically exosomes, on recipient cells16,17,18. Numerous studies have implicated EVs in the pathogenesis of a variety of infections, including HIV-16,10,12,19,20,21,22. However, the size similarity between EVs and virions presents a potential obstacle in defining these EV-mediated mechanisms. To address this concern, we have designed our protocol to implement a density gradient to effectively separate EV populations from virions. To this end, cell culture supernatant from HIV-1 infected cells were subjected to the presented protocol and analyzed by western blot for the presence of HIV-1 Gag p24 to determine which fraction or fractions contained HIV-1 virions. The results shown in Figure 3A (middle panel) shows the localization of p24 in three independent conditions: in the absence of an inducer, in the presence of an inducer (Phorbol 12-myristate 13-acetate; PMA), and in the presence of PMA and combination antiretroviral therapy (cART), the standard treatment for HIV-1. These data indicate that the HIV-1 virus is localized to fraction 16.8 as measured by the presence of p24 and its uncleaved polyprotein Pr55. Furthermore, when cells are subjected to cART, which includes Indinavir, a protease inhibitor which prevents the cleavage of Pr55 to p24, no p24 was detected in any fraction. Instead, only Pr55 was detected and was present in the 16.8-18.0 fractions, indicating a shift of virus into more dense fractions. Similar results were obtained using primary macrophages (lower panel). Although the described protocol results in the co-sedimentation of Exo #2 and Exo #3 populations with virus, the Exo #1 population (fractions 10.8-12.0) remained virus-free, allowing for downstream assays free of virus contamination.
This type of separation of virus away from EVs allows us to address the possibility of pieces of virus entering EVs or being secreted from infected cells as free protein. Results in Figure 3B indicate that HIV-1 Nef protein can be found in the Exo #1 population in addition to the Exo #2 and Exo #3 populations. Nef also appears in the 6.0 fraction, indicating that Nef can be potentially secreted from infected cells as a free protein and within an EV. Additionally, HIV-1 Vpr protein is found in the 6.0 fraction while HIV-1 Tat protein is predominantly found in the 6.0 fraction, but also appears in fractions 7.2-9.6. We are currently testing all fractions for the presence of Tat protein by ELISA. These results indicate that both Tat and Vpr can be secreted from infected cells, likely as free proteins, while Tat could also be incorporated into EVs. Collectively, these data indicate that our method of EV isolation can successfully isolate exosomes (Exo #1) away from HIV-1 virions (Exo #2 and #3) and that EVs from HIV-1-infected cells contain some HIV-1 proteins, which may affect HIV-1 pathogenesis.
Characterization of EVs and separation of EVs away from other viruses
To apply this protocol to other viral infection models, we have characterized vesicles from cells infected with several different viruses. Figure 3C shows an illustration of previously obtained vesicle and virus distributions following precipitation, separation, and enrichment according to the described protocol. As previously shown in Figure 3A, exosomes (CD9+CD63+CD81+) are present in three different populations (Exo #1, fractions 10.8-12.0; Exo #2, fractions 15.6-16.8, and Exo#3, 18.0 fraction) and HIV-1 virus localizes to the higher density 16.8 and 18.0 fractions (lanes 10-11). Not surprisingly, when applying this protocol to characterize EVs released from Human T-lymphotropic virus Type 1 (HTLV-1), a retrovirus that is known to cause cancer in adults, we observed results similar to that of HIV-1-related EVs. The third panel in Fig. 3C shows that the HTLV-1 virus was primarily localized to the highest density fraction (18.0) and, to a lesser extent, fractions 13.2-16.8 as evidenced by the presence of HTLV-1 matrix protein (p19). Furthermore, we have previously found that the HTLV-1 transactivating protein, Tax, which is absent from the HTLV-1 virion, is present within exosomes released from HTLV-1 infected cells15,23. The data in Figure 3C shows that Tax was present in the lower density fractions (6.0-12.0), two of which we have shown to contain exosomes (10.8 and 12.0).
Next, we tested the isolation protocol in the context of larger and smaller viruses such as Ebola virus (EBOV) and Zika virus (ZIKV), respectively, as HIV-1 and HTLV-1 virions are both retroviruses and very similar in diameter. Using VLP as a surrogate biosafety level 2 (BSL-2) model of EBOV assembly and exit, we found that VLP, which is approximately 1 µm in length, localizes to the highest density fraction (18.0) in the absence of 0.22 µm filtration (middle panel, Figure 3C). Moreover, when VP40, the EBOV matrix protein, is expressed at high levels, it is secreted from the cell through several different mechanisms resulting in the presence of VP40 in every density fraction, including those to which exosomes are localized13,14. In contrast, ZIKV virions are significantly smaller than HIV-1 virions, measuring approximately 40 nm in diameter. The representative diagram in the bottom panel of Figure 3C shows the presence of ZIKV virions in both low density (6.0-8.4) and high density (15.6-18.0) fractions, suggesting the presence of both free virus and EV-encapsulated virus, respectively.
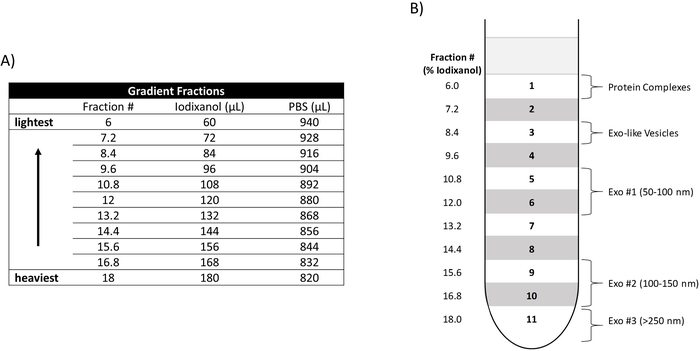
Figure 1: Construction of an iodixanol density gradient. (A) Relative amounts of iodixanol and PBS utilized to create each density fraction (Fraction #). The Fraction # denotes the percentage of iodixanol included in each fraction. Their relative densities and placement in the gradient are depicted from heaviest to lightest. (B) Placement of the density fractions (6.0-18) in ultracentrifuge tube is shown (left side) as well as the location of the three EV populations (Exo #1, Exo #2, Exo #3), Exo-like vesicles, and protein complexes. Please click here to view a larger version of this figure.
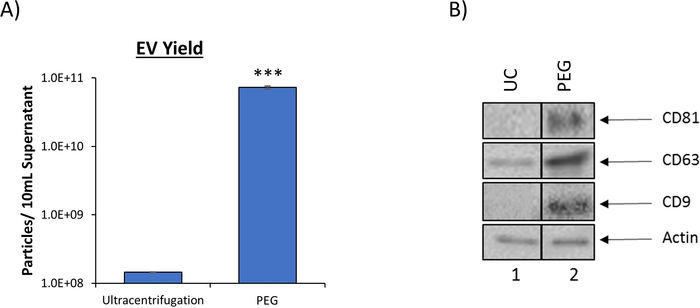
Figure 2: PEG precipitation reagent increases EV yield. (A) To compare EV recovery, EVs were isolated from 5 d HIV-1 infected monocyte (U1) culture supernatant (10 mL) using traditional ultracentrifugation (100,000 x g) or PEG precipitation reagent (incubation at a 1:1 ratio). EVs from both enrichment procedures were analyzed using nanotracking analysis to assess resulting EV concentration as described in DeMarino, et al.6. NTA was measured at 11 independent positions in technical triplicate. The blue bars represent an average of the 33 measurements ± S.D. Statistical significance was determined using a two-tailed Student’s t-test; ***p < 0.001. B. EVs from 5-day CEM culture supernatant using either ultracentrifugation (UC) or PEG precipitation followed by iodixanol density separation. Ultracentrifugation was performed using 100 mL of culture supernatant (Lane 1) while PEG precipitation and subsequent iodixanol fractionation was performed using 10 mL of culture supernatant (lane 2). The total EV pellet obtained from ultracentrifugation and the 10.8 fraction from PEG/iodixanol separation were analyzed by western blot for the presence of exosomal marker proteins (CD81, CD63, and CD9) with Actin as a control. For clarity, selected lanes from the same blot with identical exposures are shown in panel B. This figure has been modified from DeMarino, et al.6. Please click here to view a larger version of this figure.
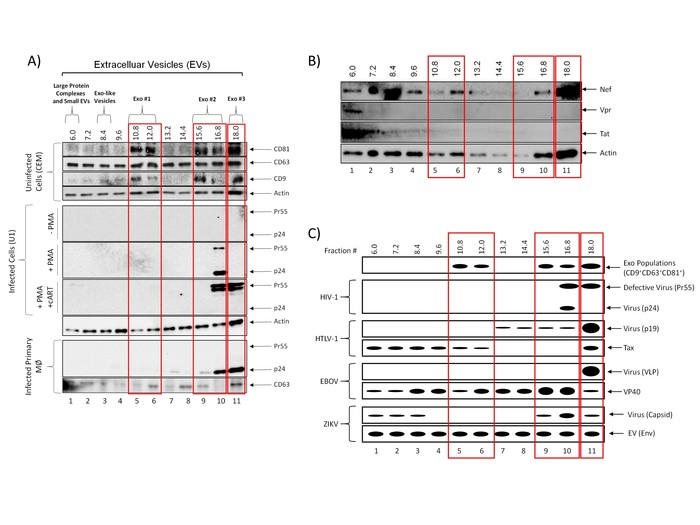
Figure 3: Isolation of EVs away from viruses. (A) Five-day culture supernatants from uninfected CEM cells (top panel), HIV-1 (Ba-L; MOI: 0.01) infected U1 cells (±PMA and ±cART treatment; middle panel) or HIV-1 infected primary macrophages (bottom panel) were incubated with PEG precipitation reagent at 4 °C overnight. EVs were separated into fractions using an iodixanol density gradient. EVs from all fractions were enriched using NT80/82 particles overnight at 4 °C. CEM nanopellets were analyzed using western blot for the presence of exosomal marker proteins CD81, CD63, and CD9. U1 (±PMA and ±cART treatment) and HIV-1 infected primary macrophages were analyzed for the presence of HIV-1 Gag protein (p24 and Pr55; cleaved and uncleaved HIV-1 Gag polyprotein, respectively), as well as CD63. Blots were probed for actin as a control. True exosome populations (CD81+CD63+CD9+) are outlined in red. This figure has been modified from DeMarino, et al.6 (B) Five-day culture supernatants from infected U1 cells were incubated with PEG precipitation reagent at 4 °C overnight. EVs were separated into fractions using an iodixanol density gradient. EVs from all fractions were enriched using NT80/82 particles overnight at 4 °C. Fractions were run on a Western blot for the presence of HIV-1 Nef, Vpr, and Tat. Actin was used as a control. True exosome populations (CD81+CD63+CD9+) are outlined in red as previously defined in DeMarino, et al.6 (C) Representative illustration of previously characterized EV separation profiles from four different viruses: HIV-1, HTLV-1, EBOV, and ZIKV. Please click here to view a larger version of this figure.
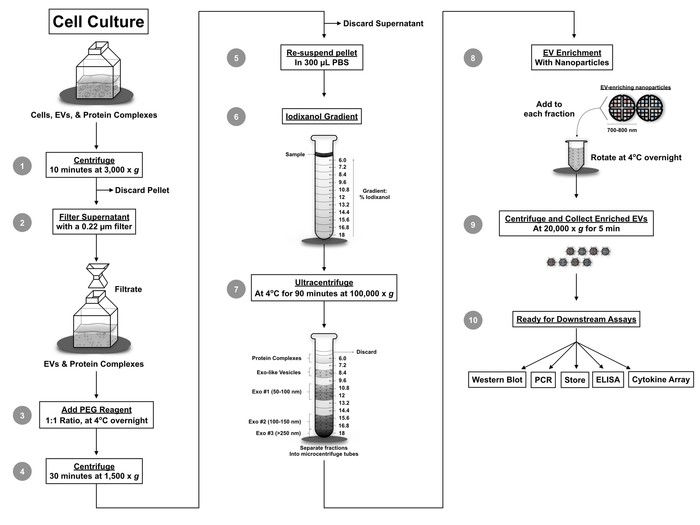
Figure 4: Protocol workflow. The novel workflow combines several techniques including filtration, EV precipitation using PEG precipitation reagent, iodixanol density gradient separation, and nanoparticle enrichment of EVs. Utilization of this protocol separates EVs from various viruses and provides a small volume sample for downstream assays including qPCR, Western blot, mass spectrometry, RNA sequencing, cytokine analysis, ELISA, and cell-based assays. This figure has been modified from DeMarino, et al.6. Please click here to view a larger version of this figure.