Performing in vitro analysis of exonuclease activity requires a number of preparatory steps in addition to the actual analysis. An overview of the procedures is shown in Figure 1.
Prior to conducting fluorescence-based exonuclease assays, it is critical to optimize detection of the fluorescently labeled oligonucleotide substrate following separation on urea-acrylamide gels using a suitable fluorescence imaging system. Filter choice is extremely important as this can have a marked impact on the sensitivity of the overall assay – our oligonucleotide substrate is fluorescein-labeled so requires a filter that permits excitation at 470 nm and emission at 520 nm. Using this filter, it is possible to increase the ability to detect low concentrations of substrate (or product) by adjusting both the sensitivity and resolution of the imager (Figures 2A and 2B). Note there is some tolerance in terms of filter bandwidth, as a suboptimal filter choice (ex 520/em 580 nm) still allows partial detection of the substrate, though with much lower sensitivity (Figure 2C). We suggest using a combination of labeled oligonucleotide concentration and imager settings that allow robust detection of substrate (Figure 2B and dark grey bars on right panel of Figure 2) so that degradation products, which are present at lower amounts in each band as the substrate is sequentially fragmented, can be reliably detected.
A successful exonuclease assay will cause degradation of the substrate sequentially towards the fluorophore (according to the polarity of the exonuclease being tested, this can be either a 5' or 3' end label on one strand of a duplex), resulting in a characteristic ladder-like pattern of DNA on the gel representative of exonuclease degradation (e.g. a time course of degradation is shown in Figure 3). Single stranded oligonucleotides can also be used but note that nucleases may show a length requirement for cleavage of ss DNA15,17 so false negatives may result from using too short an oligonucleotide. The kinetics of the enzyme activity can be approximated by performing densitometry of the gel images of a time course experiment and quantifying amount of nondegraded substrate remaining compared with amount present in smaller fragments (i.e. products of degradation – see Figure 2 of Mason et al.14 for an example). One simple method is to divide the gel equally into four vertical sections then obtain densitometry readings of each quarter. This avoids issues with minor variation in mobility of the substrate across the gel, though of course does not permit sensitive measurements such as cleavage of one or a few nucleotides from the substrate. It is important to normalize densitometry measurements against a region of the gel lacking any DNA (e.g. leave at least one lane empty), and to calculate degradation against an oligonucleotide-only control that is loaded for every experiment (see Mason et al.14).
Once optimized for detection, the assay can be used to detect differential activities both qualitatively and quantitatively. For example, the presence or absence of activity can be determined for nuclease mutants compared with wild type protein (Figure 4), while different substrates may also be tested for their ability to be degraded by the nuclease under investigation (Figure 4). If processivity measurements are required, it is possible to add excess unlabeled oligonucleotide at a defined time point and determine cleavage activity post-addition. A highly processive enzyme will continue to cleave the labeled substrate, while a poorly processive enzyme will show greatly diminished capacity to cleave the labeled substrate as it dissociates from substrate and reassociates with the unlabeled oligonucleotide that is in molar excess (e.g. see Mason et al.14 Figure 2).
Similarly, the intensity and migration of bands of the ladder will provide data on the activity and processivity of a particular exonuclease, either intrinsically or under differential conditions such as cation availability or temperature. As described previously14, DmWRNexo is partially processive but does not readily cleave the 5' fluorescein-labeled 50 nucleotide template to completion under the conditions used here. Single nucleotide bands are thus rarely seen with this enzyme and substrate. However, the assay can readily detect single nucleotide cleavage products, as observed when DmWRNexo (a 3'-5' exonuclease) acts on a 3' fluorescein-labeled substrate (see Figure 3 of Mason et al.14). Indeed, determination of directionality of exonuclease activity is achieved by analysis using substrates with backbones labeled either at the 5' or 3' end – a 3' exonuclease will immediately cleave the fluorophore from a 3'-labeled strand, rendering the subsequent laddering activity invisible to analysis, and resulting in only a single very-high mobility species apparent on the gel (see Figure 3 of Mason et al.14). Endonucleases that cut internally will show products at specific mobilities rather than a ladder. Similarly, altered bases that are not susceptible to nuclease cleavage will result in strong pause or stop sites that can be detected as more intense bands representing cessation of cleavage at those modified bases (e.g. See Figure 7 of Mason et al.14).
Nonoptimal results include smearing i.e. nonspecific degradation. Other issues to be aware of are gel problems that cause poor separation e.g. where the gel has not set evenly (Figure 5), bubbles are present in the gel, or the buffer leaks during the electrophoresis run. It is also important that substrates are checked before use as they might have separated or degraded, in which case they should be reannealed or discarded. Loss of protein activity through freeze/thaw cycles, or insufficient concentrations, can lead to negative results, though it is important to optimize for buffer requirements and other requirements e.g. ATP at high concentrations can inhibit DmWRNexo, presumably by titrating out Mg2+ ions15.
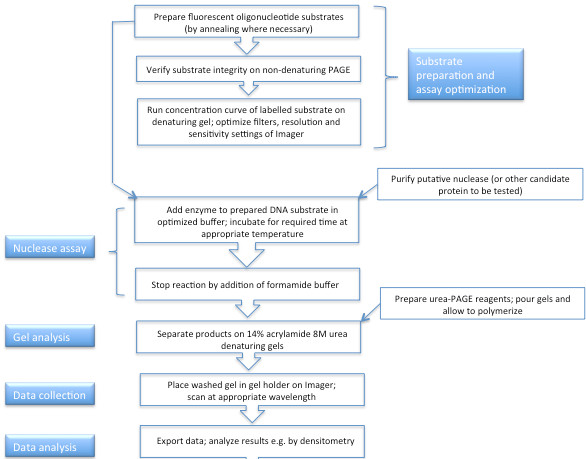
Figure 1. Flow chart of procedures undertaken when conducting a fluorescence-based exonuclease assay.
Click here to view larger image.
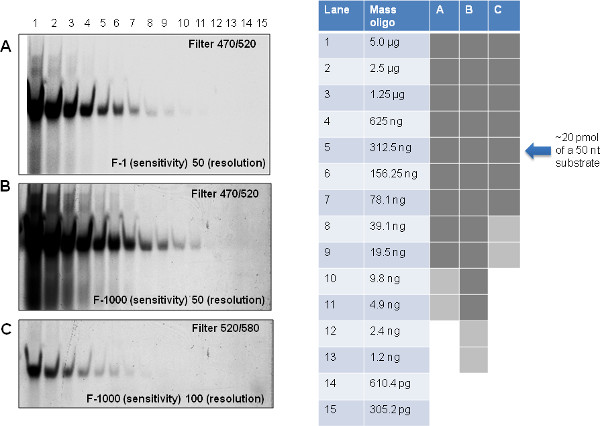
Figure 2. Optimizing detection of fluorescent substrate using the phosphorimager. A single gel of doubling dilutions of the 50-mer fluorescently labeled oligonucleotide substrate was exposed on the Phosphorimager using different filter conditions (A, B: 470 ex/520 em which is optimal for fluorescein, or C: 520 em/580 ex) and altering either sensitivity or resolution. Dark grey boxes indicate oligonucleotide concentrations that can be detected reliably with a high sensitivity and resolution; lighter grey boxes indicate those concentrations at which the signal can still be detected though the signal is much weaker. Unshaded areas of the table represent oligonucleotide concentrations that could not be detected using the filter, sensitivity and resolution settings indicated on the gel images A, B and C. Note that 20 pmol of a 50 nucleotide substrate is approximately equivalent to 312.5 ng. Click here to view larger figure.
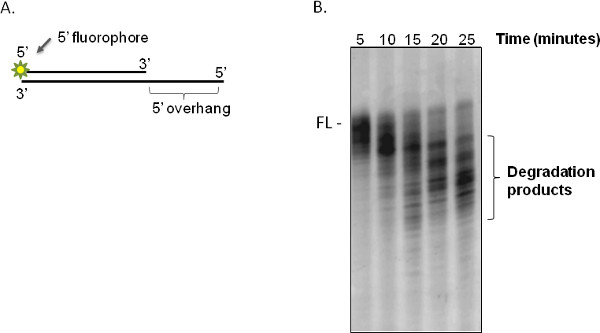
Figure 3. Typical fluorescently labeled substrate and exonuclease assay results. (A) Schematic representation of a duplex oligonucleotide substrate (5'OV) with 5' overhang on the unlabeled guide strand and 5' fluorescein label on the cleaved strand, as used in these assays. (B) Time course of degradation of the 5' overhang substrate by purified DmWRNexo at 37 °C. The enzyme binds to the 5' overhang of the guide strand and sequentially cleaves the labeled strand in a 3'-5' direction, resulting in a ladder of fragments of progressively smaller sizes that run more quickly on acrylamide gels. In this experiment, 10 pmol of DmWRNexo and 20 pmol oligonucleotide substrate were used in a 20 μl reaction. ('FL' represents the position of the nondegraded duplex 5'OV duplex fluorescent substrate).
Click here to view larger image.
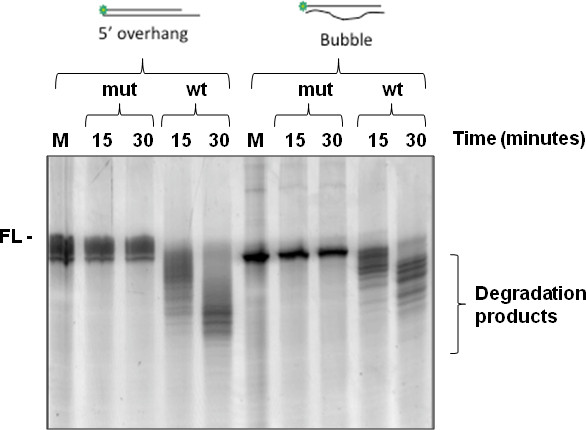
Figure 4. Testing the impact of point mutation of the exonuclease catalytic domain on ability of the enzyme DmWRNexo to cleave either duplex substrate with 5' overhang, or a bubble structure. In both cases, the substrate is labeled on one strand only at the 5' end with fluorescein. In a 20 μl reaction, 12.5 pmol purified DmWRNexo protein (wt or double point mutant D82A E84A) was incubated at 37 °C with 25 pmol oligonucleotide as shown in the schematic above the gels, and products analyzed on urea-acrylamide gels after 15 or 30 min. ('FL' represents the position of the nondegraded fluorescent substrate). 'wt' denotes wild type DmWRnexo protein, while 'mut' is a double point mutant version (D82A E84A). These results were useful in confirming the assignment of the enzyme's active site, which had been based on homology to human WRN exonuclease domain together with in silico structural modeling.
Click here to view larger image.
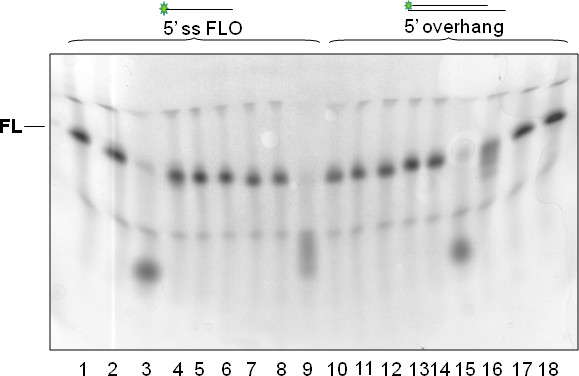
Figure 5. One example of a poor result. As a consequence of uneven polymerization of the gel, the fluorescently labeled substrate does not run at the same apparent mobility in all lanes. This can lead to incorrect interpretation of cleavage data, even though it is apparent that the substrates in lanes 3, 9 and 15 have been cleaved almost to completion, with partial cleavage in lane 16. Note that some lanes appear compressed in width in addition to the aberrant mobility leading to the 'smile' detected. Using identical conditions for polymerization (e.g. fresh APS, suitable concentrations of TEMED and a stable and consistent room temperature) can help to avoid such problems. 'FL' represents the position of the nondegraded fluorescent substrate.
Click here to view larger image.
