This study demonstrates a step-by-step protocol for studying cell death initiation through lesion occurrence on potato cv. Rywal, with a digital microscope. This enables determining the exact time of programmed cell death initiation.
Plants that have developed roots were put in soil 2 weeks after potato cv. Rywal micropropagation (Figure 1A,B). After 3-4 weeks of growth under described conditions, plants with at least 3-4 fully developed leaves with visible leaflets that looked healthy, with no signs of abscission, were used for further analysis (Figure 1C). Using a digital microscope as described in this protocol, we observed the same area on the inoculated leaf at 15-min-intervals and determined the lesion occurrence and expansion in time (Figure 3). The lesion occurred at 15 h 30 min (Figure 3).

Figure 1: Plant preparation for analysis with a digital microscope. (A) A plastic box with MS 30 medium and potato cv. Rywal plant explants containing nodes. (B) Potato cv. Rywal plant in soil (2 weeks after micropropagation). (C) Potato cv. Rywal plant, ready for inoculation (4 weeks after being put in soil), having at least three fully developed leaves. (D) Second inoculated leaf (arrow) of potato cv. Rywal plant positioned and immobilized (arrow) with tape. (E) Plant positioned under the digital microscope with the arrow pointing to the dial used for focusing. Please click here to view a larger version of this figure.
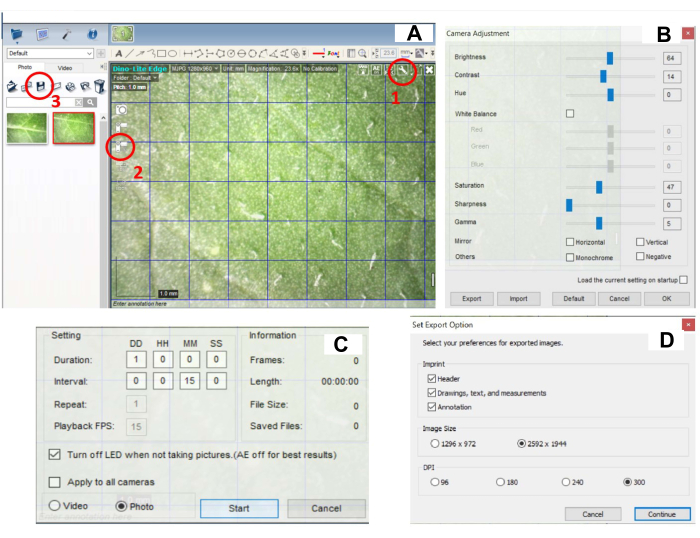
Figure 2: Digital software setting for recording lesion development. (A) Software interface – circled with red are options for the button for (1) camera settings, (2) image capture settings, and (3) saving images. (B) Window with camera settings, which opens with a click on (1) in panel A. Brightness, Contrast, Saturation, Sharpness, and Gamma should be properly adjusted. (C) Window with image capture settings, which opens with a click on (2) denoted in panel A. (D) Window with image saving settings, which opens with a click on (3) denoted in panel A. Please click here to view a larger version of this figure.

Figure 3: Lesion formation on the inoculated leaf observed under the digital microscope. Images of the central part of PVY-inoculated potato leaf at 23.6x magnification as seen under the digital microscope, taken at intervals of 5 min. Inoculated plants were put at 28 °C for 3 days, and on the third day, the observation with a digital microscope at 22 °C started at 7:00. (A) At 21:02, the lesion is not yet visible, (B) 90 min later, at 22:32, the lesion is visible. (C) The lesion expansion was observed at 01:02 and (D) 07:32 the next morning. Experiment was repeated two times, and the lesions occurred 8 h 15 min, and 12 h after cell death initiation, respectively. Please click here to view a larger version of this figure.
| Alcohol burner | Mikro+Polo | SH-234002455 | For tweezers and scalpel sterilization |
| Autoclave A-21 CAV | Kambi |
N/A | |
| Bacto Agar | Becton, Dickinson and Company | 214010 | |
| Carborundum powder | VWR Chemicals | 22505297 | |
| DinoCapture 2.0 | Dino-Lite | Version 2.0 | software for digital microscope |
| Dino-Lite Edge AM7915MZTL digital microscope | AnMo Electronics Corporation | AM7915MZTL | |
| Ethanol, 70% | Stella Tech | P94000 | For tweezers and scalpel sterilization |
| Extraction bags | Bioreba | 420100 | |
| Growth chamber FS-WI | Photon Systems Insturments | N/A | |
| Hand homogenizer | Bioreba | 400010 | |
| Hawita Special Substrate | HAWITA Gruppe | 2000000071701 | Ready to use substrate, made using peat (H4-H6 and H6-H8) |
| Hydrochloric acid (HCl) | Merck | 109057 | |
| Label tape | Sigma | L8144-5EA | |
| Laptop computer with installed DinoCapture 2.0 | HP | Z2V77EA#BED | Computer needs to be transferable as experiment takes part in a growth chamber |
| Murashige and Skoog medium | Duchefa Biochemie | M02220100 | |
| Na2HPO4 | Emsure | 1065860500 | |
| NaH2PO4 | Emsure | 1064700250 | |
| Pasteur pipette 0.5 mL | Brand | 21500209 | |
| pH-meter | Mettler Toledo | ML1601 | |
| Plastic boxes | Cvetlice Dornig | VCG10.5 | Radius = 10.5 cm |
| Plastic pots | Lab Associates | DIS40003 | Radius = 11.5 cm (top), Radius = 9.8 cm (bottom) |
| Saccharose | Kemika d.d. | 1800408 | |
| Sodium Diethyldithiocarbamate (DIECA) | Sigma-Aldeich | 228680 | Sodium diethyldithiocarbamate trihydrate, ACS reagent |
| Sodium hydroxide (NaOH) | Merck | 106462 | |
| Sterile surgical blades | Braun | 4511733633 | |
| Tweezers | Braun | BD033R |
