There are two microinjection-based methods that can be used to measure the kinetics of mRNA export in mammalian cells; injection of in vitro synthesized mRNA, or plasmid DNA, which is transcribed in vivo into mRNA. Each technique has its advantages and drawbacks. Here we describe both techniques and discuss the differences between the two approaches.
1. Preparation of materials for injection
- In vitro transcribed mRNA
- mRNA is transcribed from either plasmids or PCR products that contain the gene of interest flanked upstream by the appropriate RNA Polymerase promoter (i.e. T7 or SP6 promoters). Transcription is performed in vitro using the appropriate enzyme (T7 or SP6 RNA Polymerase, Invitrogen) with appropriate nucleotides and excess cap analogue.
- Transcripts are polyadenylated in vitro using Poly-A-Polymerase (Invitrogen) and ATP according to the manufacturer’s protocols.
- The mRNA is then purified using columns from either Invitrogen or Qiagen.
- Eluted mRNA are then precipitated by adding 1/20th volume 3M potassium acetate and 2 volumes of 100% ethanol at -20°C for one hour followed by centrifugation at 13,000g at 4°C for 30min. If excess liquid is present, the pellet can be washed with ice cold 70% ethanol.
- The resulting mRNA pellet is air dried then solubilised in injection buffer (140mM KCl and 10mM HEPES, pH 7.4). Note that any contaminant ethanol may be toxic to the microinjected cell. The solubilized mRNA can be kept at -80°C for long-term storage.
- For microinjection, mRNA is diluted to 200μg/ml in injection buffer and mixed with Oregon Green 488 (OG)-conjugated 70kDa dextran (1mg/ml; Invitrogen). Since the OG-conjugated dextran is too large to diffuse across the nuclear pore, it will enable one not only to identify injected cells, but also help to determine how much of the fluid was injected into the nucleus and how much leaked into the cytoplasm (see 4.1.2). Alternatively, any fluorescent, high molecular weight molecule that is unable to traverse the nuclear pore may be used.
- Samples are centrifuged at 13,000g at 4°C for at least 20min prior to loading the needle in order to pellet any particulate matter that may potentially obstruct the needle tip.
- Plasmid DNA
- Plasmid DNA containing the gene of interest can be prepared using standard DNA purification kits from Qiagen. Generally larger DNA preparations tend to be of higher quality and thus are more efficiently transcribed after injection.
- Plasmid DNA is diluted 50 to 200μg/ml in injection buffer containing OG-conjugated 70 kDa dextran (1mg/ml).
- Prior to loading the needle, the injection fluid is centrifuged at 13,000g at 4°C for at least 20min.
- Needles for microinjection
- Needles are fabricated from 1.0mm borosilicate glass capillary tubes (Item # 1B100F-3; World Precision Instruments Inc.) using a Sutter p97 Flaming/Brown Micropipette Puller with a 2.5mm filament.
- The injection needles are generated using a three step pulling program using a 2.5×4.5 mm box filament (item# FB245B, Sutter Instrument Co.). The program parameters used in this experiment are listed below, however they should be optimized for each filament.
Step # Heat Pull Velocity Time Pressure 1 740 100 8 250 500 2 740 100 8 250 500 3 740 100 10 250 500
(Ramp temperature=740)
- Cell preparation
- Generally any mammalian cell line can be microinjected, however cell types that are well spread tend to be more amenable to injection. The choice of cell line may also depend on other factors. For example, mRNAs that code for secreted proteins are targeted to the surface of the endoplasmic reticulum and this is much more visible in COS-7 cells as compared to NIH 3T3 fibroblasts6 (compare the localization of t-ftz-Δi mRNA in figures 3 and 4).
- Cells should be seeded on acid-washed square coverslips (25x25mm) in 30mm petridishes for at least 24hrs prior to microinjection. In certain cell lines, spreading can be stimulated by plating cells on fibronectin coated coverslips6,8. Ideally the cellular monolayer should be roughly 70-90% confluent at the time of injection.
- To allow for easy identification of injected cells, the cell monolayer is wounded prior to injection by using a 200μl plastic pipette tip. Generally a cross wound (Figure 1) is etched on the coverslip and the cells are left to recover for at least 15min prior to injection in the tissue culture incubator.
- During microinjection, tissue culture media slowly loses CO2 to diffusion and as a result becomes alkali. To help maintain a neutral pH during injection, the media can be supplemented with 10mM HEPES pH 7.4. The extra buffer can be added to the media the day before microinjection.
- The Microscope and Micromanipulator
- Cells are microinjected using an inverted microscope that is isolated on an air table to minimize vibrations that can be disruptive to the microinjection process. The microscope is equipped with two objectives; a dry 10x piece, which is used to position the cells and the needle; and a dry 40x extra long working distance phase objective, which is used to image the microinjection process.
- Optical aberrations caused by viewing cells across the coverslip and petridish can be eliminated if the objective has a correction collar. Ensuring that the microscope’s brightfield is properly aligned for Köhler illumination9, will also help correct for aberrations caused by light being scattered by the injection needle (see 2.2.4).
- The needle is controlled by a three-axis hanging joystick micromanipulation device (NT-88-V3MSH, Narishige). The coarse manipulator is secured to the back pillar of the microscope to allow the needle to be easily raised and lowered into the dish while retaining its original position.
- The needle is secured to a wand that is clamped to the coarse manipulator. A tube connects the wand to a 5cc glass syringe (Item # 512311; Becton Dickinson), which generates the injection pressure necessary to eject fluid from the tip of the microinjection needle. To control the pressure level, the syringe barrel and piston are held in position by a syringe regulator which consists of two stoppers, a pipe clamp (available at any hardware store) and two metal clamps (Figure 2). To ensure that the syringe maintains high pressure, vacuum grease (Dow Corning) is applied to the syringe plunger.
- The Fluorescent Hybridization Probe
- The primary sequence of the injected nucleic acid is folded using RNA secondary structure prediction software, such as RNAstructure 4.610.
- The mRNA folds are visually assessed to identify a region of about 50 nucleotides that tends to be free of secondary structures with high melting temperatures, such as long double strands.
- The reverse complement of this region is synthesized with an Alexa546 fluorophore attached to the 5′ end of the probe (these can be purchased from Integrated DNA Technologies).
- The probe is diluted with water to a concentration of 100μM and is stored at -80°C for 2-3 years.
2. Intranuclear Microinjection
- Loading the injection fluid on the microinjection microscope
- Approximately 1μl of injection fluid is drawn from the centrifuged DNA or mRNA sample using GELoader tips (Item # 022351656; Epindorff). Liquid should be aspirated from the top of the sample to avoid disrupting the pellet which contains particulate matter that can clog the needle.
- The tip of the pipette is inserted into the back end of the needle and the liquid is ejected. Liquid will be drawn to the needle tip by capillary action and a meniscus near the tip should be visible within 5-30sec.
- The fluid-filled needle is inserted into the wand, which is then clamped to the micromanipulator at a 45° angle.
- The needle is visualized with the 10x objective and is positioned in the center of the viewing plane near the focal plane using the coarse micromanipulator knobs. The needle is then raised a few millimeters to prevent it from being damaged in later steps (2.2.2) and then the microscope back pillar is pushed back.
- The pressure is increased by depressing the plunger 0.5-2cc.
- Microinjection
- A petridish containing a cover-slip with a wounded monolayer is placed on the viewing stage.
- The microscope back pillar is pulled forward to an upright position, causing the condenser to be aligned and the needle to enter the liquid. This should be done carefully to prevent the needle from breaking onto the coverslip (see 2.1.4).
- Using the 10x objective, the center of the cross wound is identified and the needle is positioned above the cells to be injected.
- The magnification is increased by switching to the 40x objective and the needle is lowered and centered using the fine adjustment knobs on the joystick. If the image is out of focus, one must ensure that the incident light is properly aligned (i.e. Kölher illumination9) using a Bertrand lens (see 1.5.2).
- Injection should begin at one corner of the wound cross and continued along one edge for easy identification of injected cells during visualization (see Figure 1).
- The needle is lowered to make contact with the nucleus. One can readily tell that a cell has been injected by the notable change in phase brightness that accompanies injection.
- If the fluid fills up the entire cell it may indicate that the pressure is too high and needs to be lowered.
- If no change in phase brightness is detected, this may indicate that either the microinjection pressure is not strong enough to pierce the cell membrane, or that the needle tip is clogged. This may be the result of a number of issues including: insufficient pressure, imperfections in the needle, and blockage of the needle tip with small particulate matter. Since loading each needle into the micromanipulator is time consuming, and since the injection substrate is often very valuable, it is important to maximize the percentage of needles that can be effectively used for injections. Below is a step-by step guide for trying to unblock a clogged needle tip. After each step, one should try to inject 2-3cells to assess whether obstruction has been removed:
Action Purpose and Goal 1 Adjust focus to view the length of the needle tip. To determine whether there is an obvious imperfection in the needle or whether there is a large piece of particulate blocking the tip. 2 Place the needle tip beside a floating piece of particulate in the dish. If fluid is exiting the tip it should agitate the particle without it coming into direct contact with the needle. 3 Depress the plunger to the very end of the syringe This temporary increase in pressure should clear any obstruction at the tip. After releasing the plunger, it should rise on its own accord; if it does not, this means pressure is not being built-up in the syringe and you need to tighten the connections and/or add additional vacuum grease. 4 Raise the needle out of the cell media The force of drawing the needle out of the aqueous media can help dislodge obstructions at the tip of the needle. 5 Remove the plunger from the syringe entirely and reinsert it. This additional increase in pressure may be required to clear the tip of the needle. 6 Scratch the needle tip on a clean portion of the coverslip This further agitates the particulate within the needle and helps reposition it to relieve the obstruction at the tip. Stronger scratching may chip off the end of the tip, enabling the obstruction to exit the needle. 7 Load a new needle
Another common problem is the gradual loss of pressure within the needle, requiring frequent compensation by further depressing the plunger in the syringe. Often, adding additional vacuum grease to the syringe plunger can help maintain more consistent pressure. However this problem may be due to leaks the connections between syringe and tube, the tube and the wand, or the wand and the needle. Most air leaks can be sealed using Parafilm (Fisher) or grease if necessary, although leaks between the wand and needle can only be corrected by changing the rubber gasket in the wand. - Cells along a wound edge are microinjected along the wound edge for a fixed period of time (10-15min). With practice several hundred cells can be injected during this interval.
- After microinjection, the cells are incubated at 37°C in a tissue culture incubator for the desired amount of time prior to fixation. If injecting DNA, transcription is terminated after 20min by treating cells with α-amanitin (1μg/ml; Sigma-Aldrich) dissolved in growth media. This concentration effectively inhibits transcription from microinjected plasmid (Figure 3).
- A single needle can be reused to inject multiple coverslips, although the needle needs to be replaced when one changes the type of DNA or RNA that is microinjected. Once the needle is removed from the wand is should be disposed of and not reused.
- Dead or floating cells occasionally stick to the needle tip and can interfere with microinjection. To remove this debris from the needle, raise the needle out of the liquid by pushing back the pillar and then slowly reinsert the needle back into the liquid by readjusting the pillar to the upright position.
- To obtain an accurate measurement of the mRNA nuclear export kinetics measurement, separate samples should be fixed at 0min, 15min, 30min, 60min and 120min post-RNA microinjection, or after α-amanitin treatment.
3. Cell Fixation and Staining
- Cell Fixation and Permeabilization
- At the appropriate time, the growth media is aspirated and the cells are washed twice with 2 ml PBS solution (137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 2mM KH2PO4, pH 7.4). It is important to keep the coverslips moist by minimizing the time they are not submerged in liquid.
- The cells are fixed by adding 2ml of 4% paraformaldehyde (Electron Microscope Sciences) in PBS for at least 15min at room temperature.
- The fixed samples are washed twice with PBS solution.
- The cells are permeabilized with 2ml of 0.1% Triton X-100 in PBS for at least 15 minutes at room temperature.
- The samples are washed twice with PBS solution.
- FISH Staining
- Next the samples need to be prepared for hybridization. The cells are washed twice with 1x SSC solution (150mM NaCl, 15mM sodium citrate, pH 7.0) with 25-60% formimide. For the amount of formamide, use the same concentration that is present in the hybridization solution (see 3.2.3).
- To prepare the staining chamber, the bottom of a 150mm petridish is covered with water and a piece of Parafilm is floated on the water. The water is removed by turning the dish over, thus allowing the Parafilm to adhere to the bottom of the dish. Air bubbles are manually removed.
- For each coverslip to be stained, 100μl drops of the hybridization solution (25-60% formamide, 100mg/ml dextran sulphate, 1mg/ml E. coli tRNA, 5mM vanadyl riboside complex in 1x SSC with probe diluted 1:500) are pipetted onto the parafilm. Note that the amount of formamide should be optimized for the particular FISH probe being used.
- With the help of forceps, a coverslip is removed from a 30mm petridish, then using Whatman filter paper (VWR) the back side (cell-free side) of the coverslip is dried and excess buffer is wicked from the front side without drying the fixed cells. The coverslip is then placed face down onto the FISH solution. This is repeated for each coverslip.
- After placing the staining chamber lid back on, the samples are incubated for 5-18hrs at 37°C.
- Washing and Mounting the Stained Samples
- Several washing chambers are made in the same manner as the staining chamber (see 3.2.2).
- For each coverslip, 1 ml of wash buffer (1x SCC with 25-60% formamide) is pipetted onto the parafilm of the washing chamber. Again, use the same concentration of formamide that is present in the hybridization solution (see 3.2.3).
- To remove the coverslips from the staining chamber, 1ml of wash buffer is pipetted next to the coverslip. The liquid should be drawn underneath each sample by capillary action.
- Using forceps, the coverslips are removed and each is placed onto the drop of wash buffer and incubated at room temperature for 5 min.
- Steps 3.3.2 to 3.3.4 are repeated twice more, so that each coverslip is washed a total of 3 times.
- If the RNA is to be costained with some protein by immunofluorescence, see section 3.4.
- Slides are washed with 70% ethanol, and dried with Kimwipes. For each coverslip, 10-30 μl of mounting solution with DAPI (Fluoromount G, Southern Biotech) is pipetted onto the slides. Each slide can accommodate two coverslips.
- Using forceps and Whatman filter paper, the back of the coverslips is dried and excess liquid is wicked off the front side of the coverslip without drying the samples. Each coverslip is then placed face down, onto the drop of mounting solution.
- Mounted samples can be store at 4°C.
- Immunofluorescence Staining
- Samples must be washed twice with PBS to remove formamide which can interfere with proper antibody staining.
- For each coverslip, 50μl of primary antibody solution (1.0μg/ml antibody, 0.1% TX-100, 0.1mg/ml RNAse-free BSA in PBS) is pipetted onto the parafilm of a washing chamber.
- Using forceps, coverslips are placed cell side down onto the primary antibody solution and allowed to incubate at room temperature for 30min.
- For each coverslip, two 1ml drops of PBS are pipetted onto the parafilm of a washing chamber.
- To remove the coverslips from the antibody staining solution, 1ml of PBS is pipetted next to the coverslip. The liquid should be drawn underneath each sample by capillary action.
- Using forceps, the coverslips are removed and each is placed onto the first drop of PBS for 5min and then placed onto the second drop of PBS for another 5min.
- Steps 3.4.2 to 3.4.6 are repeated using the fluorescent secondary antibody solution (1.0μg/ml antibody, 0.1% TX-100, 0.1mg/ml BSA in PBS). Note that since the injection marker and RNA are visible in the green and red channel, the secondary antibody must be conjugated to a compatible dye such as Alexa647.
- Coverslips are mounted as in 3.3.7.
4. Imaging and Quantification
- Imaging
- An epifluorescence microscope is used to image the cells following injection. Injected cells can be located by locating the cross wounds and identifying cells with injected marker (OG-dextran).
- For each cell observed, a picture of the injected OG-dextran is acquired. When imaging microinjected mRNA it is important that quantification is limited to cells that received >90% of the injected fluid (i.e. dextran) into the nucleus.
- An image of the RNA is then acquired. To allow for the accurate measurement of RNA, the exposure time between all cells within a given time course should remain constant and fall within the dynamic range of the camera. Ideally each image should include an uninjected cell to be able to proper calculate the background fluorescence intensity (see 4.2.4).
- To aid in quantification, an image of the DAPI stain can also be acquired. Also if immunofluorescence was performed, the stained protein can also be imaged. An example of mRNA distribution at various points in a time course is shown in Figure 4. The mRNA encodes a secreted protein and is thus targeted to the ER in COS-7 cells. Note that the ER-targeting was verified by co-staining the RNA with antibodies against TRAPα, a resident ER protein11.
- Quantification
This can be accomplished with a number of different image analysis software packages. ImageJ software is well-suited for quantifying cellular mRNA distribution since it can be used to manually separate nuclear and cytoplasmic fractions, it can measure the average fluorescence of these fractions, and its output data can be readily copied and pasted into other software, such as Microsoft Excel.- Images corresponding to FISH, OG-dextran, and DAPI fluorescence are opened and merged using the Images to Stack tool.
- In either the DAPI or dextran layer the Threshold tool is used to isolate the area corresponding to the microinjected nucleus. This fraction is selected using the Wand tool, and after moving to the FISH layer, the area (ANuc) and mean/average intensity (FNuc) are recorded using the Measure tool.
- The cell perimeter is outlined using the Freehand selection tool, and while on the FISH layer the area (ATot) and mean/average intensity (FTot) are recorded using the Measure tool. If your cell’s fluorescence is significantly more intense than that of the background, you may use the Threshold tool instead of the Freehand selection tool to outline your desired cell.
- Using the Rectangular selection function, a box is drawn over an uninjected cell and the mean/average intensity (FIndietro) is recorded.
- All measurements are copied to an excel worksheet.
- Exported mRNA is calculated using the following equation:
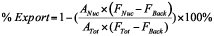
ANuc Area of the nucleus ATot Area of the whole cell FNuc Average fluorescence of the nuclear fraction FTot Average fluorescence of the whole cell fraction FIndietro Average fluorescence of an untransfected cell
For each time point the average %Export is calculated and plotted over time. - mRNA Stability is calculated by plotting the average total mRNA fluorescence (ATot x (FTot – FIndietro)) over time. Typically we find that amount of mRNA varies greatly between cells and between coverslips. This may be due to inconsistencies between needle flow rates and between the efficiency of FISH staining.
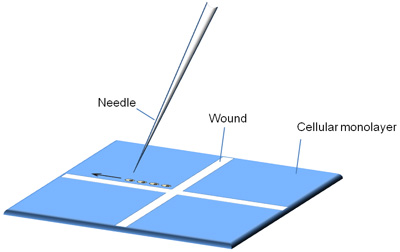
Figure 1. Microinjection coverslip. Cells are grown until they are 70-90% confluent then wounded vertically and horizontally using a 200μl plastic pipette tip. Starting from the cross-wound, cells are injected along one wound edge (arrow).
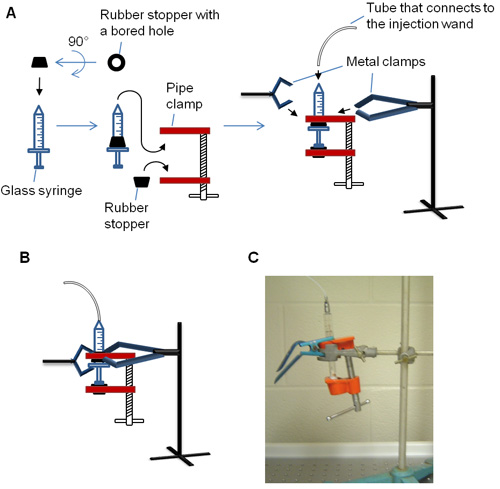
Figure 2. Syringe regulator. A) Flow diagram demonstrating how the syringe regulator is assembled. B) Schematic of the assembled apparatus. C) A photograph of the assembled syringe regulator.
Figure 3. Effects of α-amanitin on transcription of injected plasmid DNA. NIH3T3 fibroblast cells, which were pre-treated with varying concentrations of α-amanitin, were injected with t-ftz-Δi plasmid DNA and OG-conjugated 70kD dextran. Injected cells were incubated at 37°C for 1 hour and then fixed and stained for t-ftz-Δi mRNA using a specific FISH probe6. Each row corresponds to a single field of view imaged for OG-70kDa dextran and t-ftz-Δi mRNA. Note that high, but not low, concentrations of drug totally inhibited production of t-ftz-Δi transcript. Scale bar = 15μm.
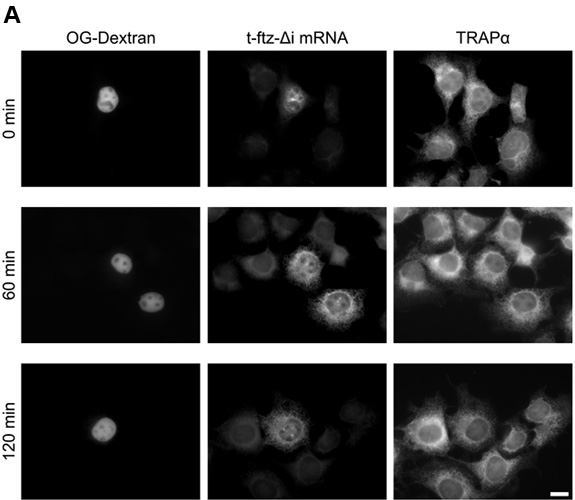
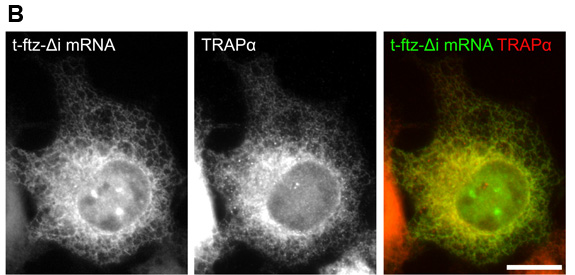
Figure 4. Time course of mRNA nuclear export. COS-7 cells were injected with t-ftz-Δi plasmid DNA and OG-conjugated 70kD dextran. 30min following injection cells were treated with α-amanitin and incubated at 37°C for the indicated time points. The cells were then fixed and stained with probe against t-ftz-Δi mRNA and with antibodies against the ER marker TRAPα. Each row corresponds to a single field of cells. A) mRNA distribution following a microinjection timecourse. B) A blow up of the 120min time point from (A) demonstrating the co-localization of t-ftz-Δi mRNA and the ER. An overlay of the t-ftz-Δi mRNA (green) and TRAPα (red) staining is shown in the right panel. Scale bars = 15μm.
