1. Establishment of fibroblast cultures from skin explants
- 4 mm punch biopsies acquired from human forearm are placed in MEM supplemented 100 units / ml penicillin, 100 μg / ml streptomycin, and 0.25 μg / ml Fungizone.
- Trim any subcutaneous fat and finely chop the biopsy into small pieces by rocking a number 24 scalpel blade against the bottom of the petri dish.
- Place tissue slurry in a 25 cm2 tissue culture flask with primary fibroblast growth media (MEM supplemented with 10% FCS, 100 units / ml penicillin, 100 μg / ml streptomycin, and 25 μg / ml Fungizone) added until the surface of flask is covered but liquid depth is insufficient for tissue to float.
- Following 3 days incubation at 37°C in a humidified atmosphere of 5% CO2 , add 3 ml of primary fibroblast growth media.
- Following another 3 days incubation change media with fresh primary fibroblast growth media, or if cells are nearly confluent they may be split 1:4 into fresh media. (Fungizone can be omitted from this point onwards).
2. Stage I – Preparation of collagen I from rat tails
Note: Protocol for approximately 12-14 adolescent (fresh or frozen) rat tails
- Prepare rat tail by washing in 70% ethanol and remove tendons as follows:
- Remove skin from rat tail by slicing in the middle of the tail from top to bottom with a scalpel and pealing along the length of the tail.
- Detach tendon from the core of the proximal region of the tail.
- Remove tendon toward the distal region of the tail avoiding the sheath using toothed forceps.
- Extract 1 g of tendon / 250 ml of 0.5 M acetic acid by stirring at 4°C for 48 h.
- Centrifuge the extract (7,500 x g) for 30 mins and discard the pellet.
- Add an equal volume of 10% (w/v) NaCl to the supernatant, and stir for 30-60 min.
- Centrifuge (10,000 x g) for 30 mins, discard the supernatant, and re-dissolve the precipitate in 0.25 M acetic acid at ˜ 1:1 ratio by stirring for 24 h at 4°C.
- Dialyze the collagen solution against 6-8 changes of 6L ˜17.5 mM acetic acid (1 ml glacial acetic acid per liter of cold water, change 2 x daily).
- Centrifuge dialysed collagen at 30,000 x g for 1.5 hrs.
- Remove supernatant and place in sterile flask.
- Adjust the collagen I concentration to ˜2 mg/ml using 0.5 mM acetic acid at 4°C.
3. Stage II – Setting up the 3D matrix with embedded fibroblasts (allow to contract for 8 days).
- Assemble the mix using cold reagents at 4°C in a pre-chilled bottle. Keep collagen I on ice.
- For one T75 flask of confluent primary fibroblasts (˜ cell number 1 x 106) use:
- 25 ml rat tail collagen (approx conc 2mg/ml)
- 3 ml 10xMEM
- 0.22 M NaOH: first, add 2ml, then drop-wise whilst stirring, until the collagen turns orange,
- but not pink (usually up to 3 ml). Approximate pH = 7.2.
- Note: It is important to ensure that the medium/gel remains neutral or slightly acidic as fibroblasts will not contract the gel if exposed to even slightly alkaline conditions.
- Trypsinise the fibroblasts, spin at 400 x g for 5 mins and remove supernatant.
- During 5 mins spin above: Prepare 12 x 35 mm plastic dishes – normally achieve 12 dishes from one flask of fibroblasts/collagen mix.
- Re-suspend fibroblasts in 3 ml FCS and immediately add to the collagen mix and stir.
- Plate approximately 2.5 ml of collagen/fibroblast per dish as quickly as possible. Try to avoid bubbles and collagen setting.
- Let the collagen set at 37°C in a humidified atmosphere of 5% CO2 in air for 10 mins.
- Add 1ml of fibroblast growth media (DMEM+ 10% FCS).
- Detach the collagen/fibroblast matrix from the sides of the dish using a pipette.
- Next day: Add 1ml of fibroblast growth media (DMEM + 10% FCS).
- Change media every other day. Allow collagen/fibroblast matrix to contract for approximately 8 days, until they fit in a 24-well dish (Contraction from ˜3.5 cm to 1.5 cm in diameter, see Figure 1).
Note: The collagen concentration should be adjusted to suit the application. The more dilute the collagen solution, the faster it will be contracted by the fibroblasts. Slight differences in contraction rate will be experienced with different batches of collagen at the same concentration. Similarly, different fibroblast cultures will contract the collagen gels at different rates, and the more fibroblasts present, the faster the rate of contraction. Collagen concentration and fibroblast density can therefore by adjusted to modify the rate of contraction, and the final density of the collagen gel.
4. Stage II – Plating cells of interest on top of the matrix
Note: Sterilize all forceps and equipment with ethanol before use.
- Using blunt forceps, gently move contracted matrix to 24-well dish. Make sure it does not fold.
- Prepare suspension of cells of interest at approximately 4 x 104 /ml and plate 1ml (after trypsinising, spin cells to get rid of trypsin) on top of the matrix. Actual number of cells needed will vary depending on cell type used. Cell medium should be normal growth medium for the cells of interest.
- Allow cells to grow to confluence on top of the matrix, approximately 3-5 days.
5. Stage III – Transferring the matrix to a grid for invasion (approximately 0-21 days)
- Cut stainless steel grids to create a tripod and autoclave prior to use (See Figure 2).
- Place sterile grid in 6 cm dish, add growth media to a level above the grid (approx 10.5ml). Place matrix on the grid and gently aspirate media so the bottom of the matrix is in contact with media but not submerged. This is referred to as the air/liquid interface, which creates a gradient that promotes invasion. Replace medium every two days (See Figure 2).
- Live cell, contextual imaging using fluorescent cells can be imaged at this stage or earlier. The contracted or fibrillar collagen I can be imaged using second harmonic generation (SHG, See figure 3). Using multi-photon excitation combined with wide-field detection we find that SHG can be imaged at least 100 μm into the matrix, whereas cells expressing cytoplasmic GFP can be imaged at least 200 μm deep.
Note: With respect to quantification of invasion, the day on which matricies are placed on the grids defines Day 0. Placement on grids generates a gradient of cell culture media that promotes invasion into the matrix. Samples can be imaged over the next 1 – 21 days (or longer) to assess biological processes such as invasion, proliferation, survival or differentiation (See reference list).
6. Stage IV – Fixation
- Add 5 ml of 4% PFA in a Falcon tube.
- Transfer the matrix onto a flat surface. Cut the matrix in half with a fresh clean scalpel (as you will be staining the cross section), lift the matrix with scalpel and transfer to 4% PFA, fix over night.
- Organotypic matrices are now ready to stain with antibody/stain of choice (See Figure 3).
7. Representative results:

Figure 1 Example of fibroblast-contracted collagen I. Mixture of fibroblast and rat tail collagen detached from the petri dish and allowed to contract over 6-8 days.
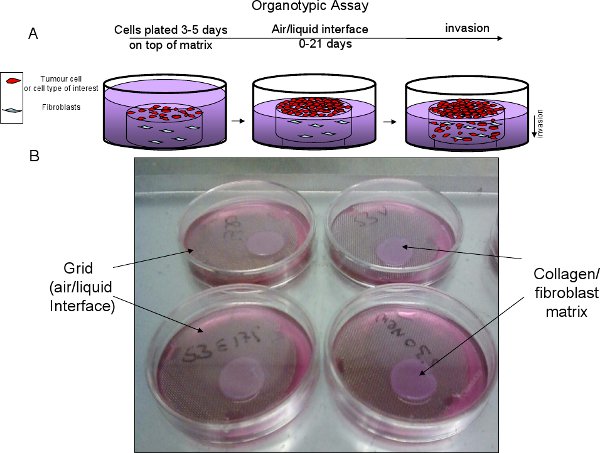
Figure 2 Organotypic assay progression. A, schematic of organotypic set up and progression. B, Example of collagen/fibroblast matrix on top of stainless steel, sterile grid to create air/liquid interface.
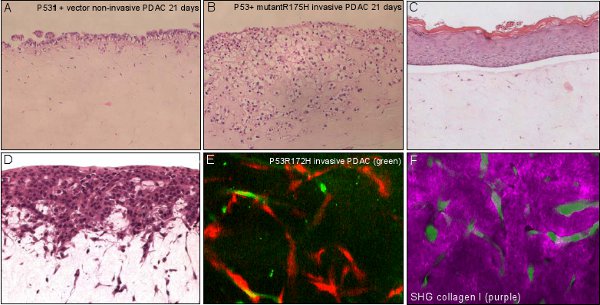
Figure 3 Applications of organotypic assay. A, B non-invasive and invasive pancreatic ductal adenocarcinoma (PDAC) cells invading over time with organotypic matrix. C, living skin equivalent showing a stratified epidermis on a fibroblast-contracted collagenous dermal component. D, C8161 melanoma cells invading in to a fibroblast-contracted collagenous dermal equivalent. E, invasive PDAC cells (green) interacting and invading with fibroblasts (red) within the organotypic matrix. F, invasive PDAC cells (green) interacting with a degrading surrounding extracellular matrix (purple) visualised using multiphoton -based second harmonic generation.
