1. Specimen preparation: Golgi-Cox
- This protocol closely follows the protocol of Mayerich et al.7.
- The C57BL/6J mouse is deeply anesthetized using isoflurane inhalant anesthesia and then decapitated.
- The brain is removed and placed into a Golgi-Cox fixation solution (1% potassium chromate, 1% potassium dichromate, and 1% mercuric chloride in deionized water).
- The brain is left in the Golgi-Cox solution in the dark, at room temperature, for 10 to 16 weeks.
- The brains are rinsed in deionized water overnight in the dark.
- The rinsed brain is immersed in 5% ammonium hydroxide solution in deionized water for 7 to 10 days in the dark at room temperature. This lengthy preparation time was to ensure infiltration of the entire brain, so that the black precipitate had a chance to completely form in the tissue.
- The brain is rinsed again in deionized water at room temperature for 4 hours and then dehydrated through a graded series of ethyl alcohols: 50% and 70% in the refrigerator (4 °C), and 85%, 95% (3 changes), and 100% (4 to 5 changes) in room temperature. The brain is left in each solution for 24 hours.
- The dehydrated brain is then put in acetone (3 to 4 changes, each for one day), followed by an araldite-acetone mixture with araldite-to-acetone ratio of 1:2, 1:1, 2:1, and finally in 100% araldite (3 changes, each time overnight), in the refrigerator (4 °C).
- Finally, the treated brain is embedded in 100% araldite and heated to 60 °C for 3 days (cf. embedding protocol in Abbott and Sotelo2).
If specimens are embedded in LR White then the specimens are transferred from the last 100% ethanol solution through three changes of LR White solution that are each kept in the refrigerator overnight, which is a standard procedure prior to polymerization. Three changes are done to ensure full infiltration. The specimen is then transferred to fresh LR White that is polymerized in a closed container at 60 °C for 24 hours, which is the required amount of time and temperature for proper polymerization. - Fig. 1 shows a Golgi-Cox stained brain embedded in Araldite.
- The cured specimen block is then mounted on the metal specimen ring using epoxy, and the sides of the block trimmed as necessary (see Fig. 2).
2. Specimen preparation: Nissl
- This protocol closely follows the protocol of Mayerich et al.7
- The mouse is deeply anaesthetized using ketamine and xylazine injected intraperitoneally and then perfused transcardially using 50 mL of room temperature phosphate-buffered saline (pH 7.4), followed by 250 mL of cold 4% phosphate-buffered paraformaldehyde (pH 7.4).
- The mouse is then perfused with 100 mL of a solution of 0.1% thionin dye in deionized water, and the body placed in the refrigerator (4 °C) for 24 hours.
- After 24 hours, the brain is removed from the calvaria and placed in a fresh solution of 0.1% thionin and left at 4 °C for 7 days. The lengthy duration of this procedure was to ensure full infiltration of thionin into the en bloc preparation.
- The brain is then destained and dehydrated through a graded series of ethanols starting with 50% ethanol and water and increasing to 100% ethanol over a time period of 6 weeks. The ethanol changes from 50% to 70% are stored in the refrigerator (4 °C) and all changes of ethanol above 70% are stored at room temperature.
- After three changes of acetone (2-4 days in each solution at room temperature), the brain is then embedded in araldite plastic following the protocol in 1.8-1.9 above. If LR white is the embedding medium then the protocol in 1.10 above is followed.
3. Specimen preparation: India ink
- The mouse is deeply anaesthetized using ketamine and xylazine (1.7 mg ketamine / 0.26 mg xylazine per 20 grams body weight) injected intraperitoneally and then perfused transcardially using 50 mL of room temperature phosphate-buffered saline (pH 7.4), followed by 250 mL of room temperature 10% neutral buffered formalin (pH 7.4). and finally with 3.0 cc of undiluted India ink.
- Whole body perfusion with saline and fixative is necessary to clear the blood from the cardiovascular system and to fix the tissues. Perfusion with India ink is necessary to completely fill the vasculature of the cardiovascular system.
- The resulting brain is then dehydrated through a series of graded ethyl alcohols (25%-100%) and then embedded in araldite plastic following the protocol in 1.8-1.9 above. If LR white is the embedding medium then the protocol in 1.10 above is followed.
4. Specimen preparation: Generic species, generic organs
- For the generic case, fixation can be done with either 10% neutral buffered formalin or 4% paraformaldehyde.
- For small tissue volumes, diffusion instead of perfusion is recommended for fixation and staining. In the case of small organisms or organs, staining also can be done during the dehydration process.
- The embedding protocol is the same as above, although the times for solution infiltration can be reduced for organs or organisms that are much smaller than whole mouse brain.
5. KESM setup and imaging
- Turn off pump, turn on stage, turn on camera, turn on illuminator.
- Raise knife and objective.
- Install specimen ring.
- Measure specimen dimension and create a configuration file for the KESM Stage Controller2 application.
- Start KESM Stage Controller2 and initialize stage.
- Lower knife/objective, turn on pump.
- Focus objective and camera, by turning the focusing knob on the optical train and adjusting the camera’s field of view relative to the knife edge.
- Press on [Go] to initiate imaging.
- See Figs. 3-8. See 9, 7 for technical details.
6. Image processing and data preparation
- Copy the KESM Stack Processor executable into the data folder under the configuration file folder (00000, 00001, etc.).
- Run the KESM Stack Processor to remove lighting artifact and normalize the background intensity in all images. Repeat for all data subfolders.
- Prepare multiscale tiles from the data set and upload and set up the files in the KESM Brain Atlas web server.
- See Fig. 9.
7. Data visualization and analysis
- Visualization using the KESM Brain Atlas. Go to http://kesm.cs.tamu.edu and select the appropriate atlas.
- Navigate as in Google maps.
- Zoom in and out as in Google maps.
- To go to a different depth, select how deep to go at each step from the pull-down menu, and then click on either [+] or [-] to increase or decrease z depth.
- Adjust the overlay number by using the pull-down menu.
- Adjust the overlay interval by using the pull-down menu.
- See Fig. 11.
- Visualization using MeVisLab.
- Launch MeVisLab.
- Create a new project.
- In the following, new modules can be creating by typing in the module name in the command box.
- Create module [Compose3Dfrom2DFiles], and go to the desired folder. Enter file string and click in [GetFileList]. Make sure the selected images can fit in memory. Click [Create3D] to create volume.
- Create module [SetWorldMatrix], and set the “Scale” under “Elementary Transforms” x, y, z to the appropriate voxel size (0.6, 0.7, 1.0 for a 10X 0.45NA objective). Link [Compose3Dfrom2DFiles] to [SetWorldMatrix].
- Set Matrix and Transforms under “Matrix Composition” to “Ignore”, “Ignore”, “Forward”.
- Create module [Arithmetic1] and set the “Function” to “Invert(Max-Img)”.
Link [SetWorldMatrix] to [Arithmetic1]. - Create module [View3D] and connect [Arithmetic1].
- In View3D, while right mouse button is pressed, move the mouse around to adjust the threshold. You can crop regions, change orientation (move mouse while left mouse button is pressed), change illumination (volume rendering or MIP), turn on/off background, etc.
- See Fig. 10.
8. Representative Results:
Here, we present whole-brain data and details. Figs. 12-15 show whole-brain India Ink, Golgi, and Nissl data sets 1, 4, 3.

Figure 1. Fixed, stained, and embedded mouse brain

Figure 2. Embedded mouse brain specimen mounted on the specimen ring.

Figure 3. The Knife Edge Scanning Microscope (KESM). A photo of the KESM is shown with its major components marked: (1) high-speed line-scan camera, (2) microscope objective, (3) diamond knife assembly and light collimator, (4) specimen tank (for water immersion imaging), (5) three-axis precision air-bearing stage, (6) white-light microscope illuminator, (7) water pump (in the back) for the removal of sectioned tissue, (8) PC server for stage control and image acquisition, (9) granite base, and (10) granite bridge.
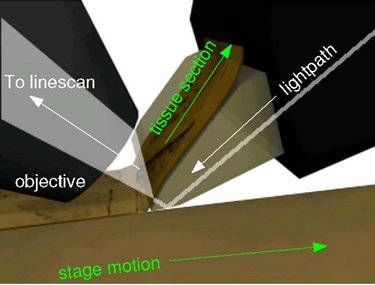
Figure 4. Imaging principles of the KESM. The principal of operation of KESM is illustrated. The objective and the knife is held in place, while the specimen affixed on the positioning stage moves (arrow with solid line) at the resolution of 20 nm and travel speed of 1-5, and gets scraped against the diamond knife (5 mm wide for 10X objective), generating a thin section flowing over the knife (arrow with solid line). Line-scan imaging is done near the very tip of the knife.

Figure 5. KESM camera and objective focusing controls.

Figure 6. KESM knife assembly and controls.

Figure 7. Initial focusing through the observation port.
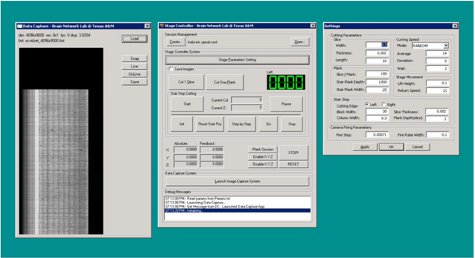
Figure 8. KESM Stage Controller 2 application (screenshot).
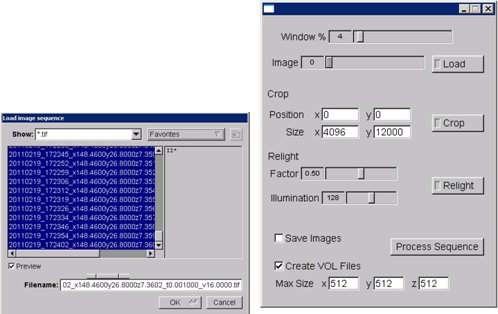
Figure 9. KESM stack processor application (screenshot).
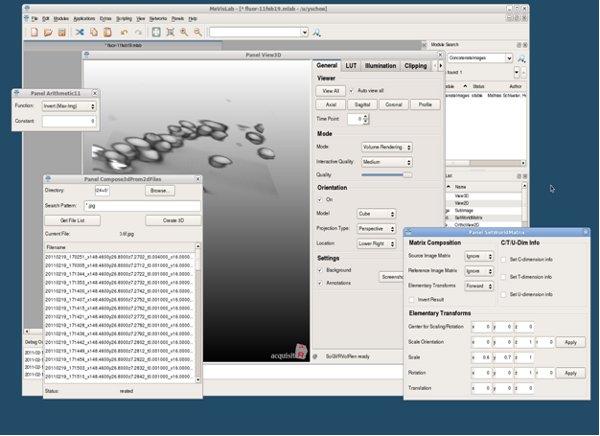
Figure 10. MeVisLab application (screenshot).
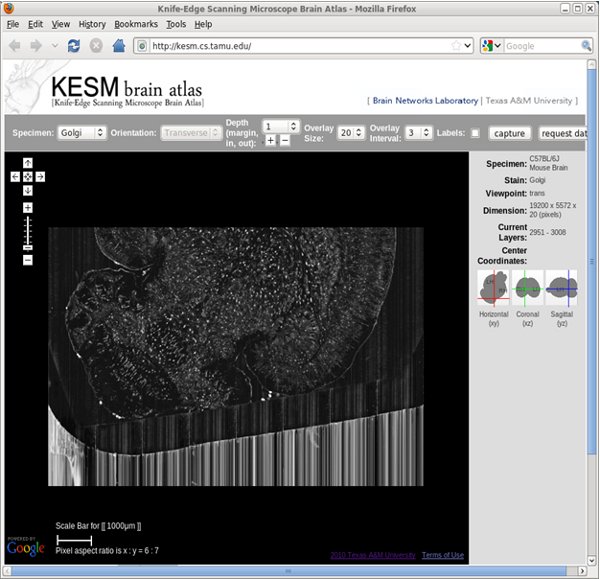
Figure 11. KESM Brain Atlas: Web-interface (screenshot).
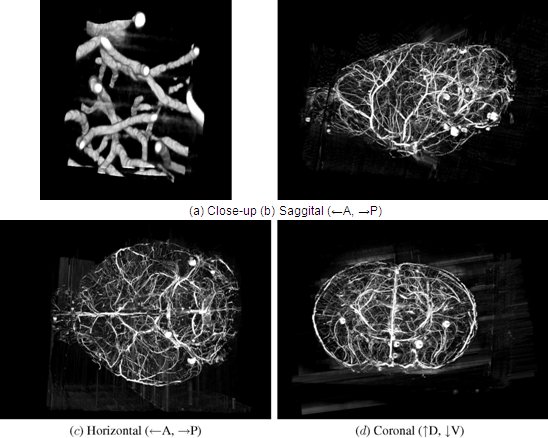
Figure 12. KESM whole-brain India ink data. Volume visualizations of KESM data stacks are shown for the vascular data set. (a) Close-up of the vascular data. Width ~100 um. (b-d) Three standard views of the whole mouse brain vasculature (subsampled from high-resolution data). Width ~10mm.

Figure 13. KESM whole-brain mouse Golgi data.
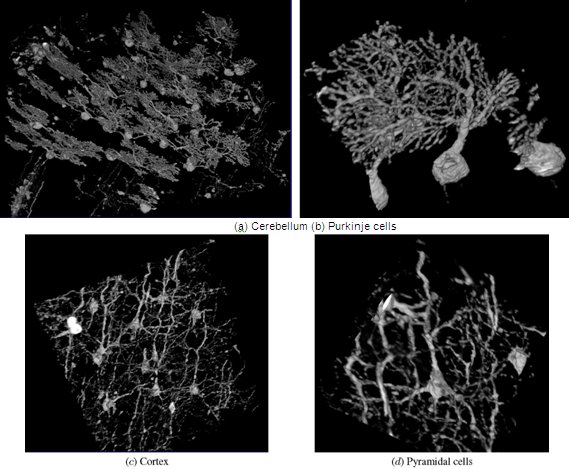
Figure 14. KESM Golgi data details.
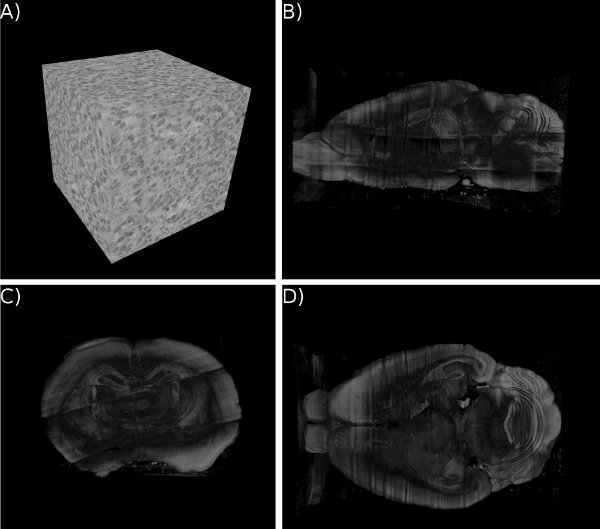
Figure 15. KESM whole-brain Nissl data. (a) Close-up of the Nissl data. ~300 um3. (b-d) Three standard views of the whole mouse brain Nissl data (subsampled from high-resolution data). Cross section shown for a clear view of the internal structures.
