The protocol as described below is divided into steps on five successive days (Fig. 1). As noted, certain steps can be done on different days. The duration of each step, as well as the quantity of material required (e.g. worms, bacteria, media) will depend on the number of 96 well plates treated per experiment.
Day 1
1. Preparation of the 96-well NGM RNAi plates
The following protocol is adapted from the standard method for making worm culture plates 10, which includes details of sterilization techniques for the different solutions.
- For 10-12 96-well plates, prepare 100 mL of Nematode Growth Media (NGM) in deionized H2O: 1.7 g BactoAgar, 0.29 g NaCl, 0.25 g Peptone, 100 μL cholesterol (5 mg/mL in EtOH).
- Autoclave NGM (5 min at 121°C for 100 mL; 30 min at 121°C for 4 L) and let it cool until it is at about 50°C (just cool enough to hold). It is important that the NGM stays warm enough so that it does not solidify.
- Add for 100 mL: 2.5 mL Phosphate buffer pH6 (1M), 100 μL MgSO4 (1M), 100 μL CaCl2 (1M), 400 μL IPTG (1M), 100 μL Ampicillin (100 mg/mL), 100 μL Tetracycline (12.5 mg/mL in EtOH).
- Distribute 75 μL of NGM to each well of a 96-well flat-bottom plate. To avoid solidification of the medium, it needs to be dispensed rapidly; it is convenient to use a repetitive dispenser (e.g. Repeatman, Eppendorf). Be sure that there are no air bubbles present in the wells.
- Store the plates immediately in a humid chamber (e.g. a Tupperware box with wet paper towels at the bottom) at 4°C.
Note: plates can be prepared up to a week in advance and stored at 4°C; this can make the organization of subsequent steps easier.
2. Automated preparation of the 96-DeepWell LB plates
- Prepare and distribute 1.5 mL LB containing Ampicillin 100μg/mL and Tetracycline 12.5 μg/mL to each well of a 96-DeepWell plate which has a maximum capacity of 2 mL/well. Distribution is optimized using a robotic liquid handling system (e.g. TECAN) but can also be performed manually if necessary.
- Add a cover to each plate and store at 4°C. In order to track each 96-well plate, a unique label or barcode should be added, at this stage or subsequently.
Note: plates can be prepared 2 to 3 days in advance and stored at 4°C; this can make the organization of subsequent steps easier.
3. Grow the RNAi bacterial clones in the 96-DeepWell LB plates (overnight culture)
- For subsequent ease of handling, when the original RNAi library clones are in a 384-well format, first redistribute the clones into labeled or bar-coded standard 96-well plates containing LB with 100 μg/mL Amp and 12.5 μg/mL Tet supplemented with glycerol (10% final concentration). When the original 384-well plates do not have clones in all wells, as is the case for the most commonly used “Ahringer” library, there are several options for organizing the daughter plates. Clones can be re-distributed so that there are no empty wells in the daughter plates. This minimizes the number of plates to screen. Ideally, however, during replication, a few wells on each daughter plate should be left empty, so that these can subsequently be filled with standard control clones (positive and negative) that facilitate across-plate comparisons. These 96-well daughter plates are then used to seed the 96-DeepWell LB plates for an overnight (ON) culture.
All liquid handling steps are best done with a robotic liquid handling system (e.g. Tecan), but can be performed manually using, for example, a 96-pin replicator. If the RNAi library clones are already in a 96-well format go to step 3.5.
- Incubate the 96-well RNAi daughter plates at 37°C with agitation (200 rpm) for 14-15 hours.
- Verify that the bacteria grew correctly and identify clones that did not grow. These will be excluded from later analyses. Ideally, the OD600 is measured in each well, but this can be done by simple inspection.
- Store the 96-well RNAi daughter plates at -80°C before use.
- Thaw the 96-well plates containing the RNAi clones at room temperature. The plates can then be kept at 4°C until use.
- Distribute 3 μL of each bacterial clone from a 96-well replicate plate to the 96 wells of the correspondingly labeled/barcoded 96-DeepWell LB plate containing 1.5 mL of LB with 100 μg/mL Amp and 12.5 μg/mL Tet. Bacterial clones to control RNAi efficiency, such as a gfp(RNAi) clone in the present example, and negative controls, can be incorporated manually in any available empty well.
- Cover the 96-DeepWell plates with an adhesive film (e.g. AeraSeal cellular culture film from Dutscher). Replace the used 96-well replicate plate at -80°C.
- The 96-DeepWell labeled plates are then incubated ON at 37°C with agitation (200 rpm for 14-15 hours).
Day 2
4. Seed the RNAi bacterial clones onto the 96-well NGM RNAi plates
This step should be done in the morning following the ON culture.
- Take the 96-well NGM plates from 4°C and let them warm up and dry for 5-15 min under a sterile laminar flow cabinet. Do not leave the plates too long under the hood (maximum 30 min), as otherwise the NGM may subsequently crack. Add the appropriate label/barcode to each plate.
- Retrieve the 96-DeepWell ON culture plates from 37°C. Record the positions of any empty wells (an empty well is completely translucent); these will be excluded from subsequent analyses.
- Centrifuge the 96-DeepWell plates 5 min at 4000 rpm.
- Tip off the supernatant by turning the plate sharply upside down and dry the edges of the plate rapidly on a paper towel. As the cultures are of recombinant bacteria, the supernatant needs to be treated in accordance with local regulations (e.g. autoclaved).
- Resuspend the bacterial pellet in the residual liquid (approximately 50 μL) by vortexing, ideally using a dedicated agitator (e.g. Tecan) so that each plate receives precisely the same treatment. It is possible to measure the OD600 of each well to standardize precisely the bacterial inoculum for the subsequent step, but this is not indispensable, and we do not perform such a check.
- Transfer 5 μL of the resuspended bacteria onto the 96-well NGM plates using an 8 or 12-channel Multi-Pipette. Be careful not to touch or penetrate the NGM. This step is performed manually, but can be optimized using a robot such as the Liquidator96 (Rainin) that allows the distribution of 96 RNAi clones in one step.
- Let the bacteria dry under a sterile laminar flow cabinet, checking regularly to avoid the NGM becoming too dry. This step should take about 2 hours.
- Incubate the 96-well RNAi-NGM plates at 37°C ON in a humid chamber.
5. Prepare a synchronized population of worms
For this step, we use transgenic worms carrying the reporter gene(s) of interest (e.g. an infection-inducible pnlp-29::GFP construct and, as an internal control, a constitutive pcol-12::dsRed transgene construct). Because of the observed decrease of transgene expression with time, we thaw fresh batches of worms every 6 weeks. We culture worms for at least 2 generations before use, and ensure that the worms have never starved before the assay. If day 2 of the protocol is Tuesday, we prepare worms on Friday by placing 30 young adult worms on a 9 cm NGM plate spread with OP50 bacteria at 20°C; they will be ready to bleach on Tuesday (see Table 1).
- Bleach worms following the standard protocol 10.
- Let the eggs hatch in M9 at 25°C ON with agitation to get a synchronized population of L1 larvae.
Day 3
6. Distribute the worms on the 96-well RNAi-NGM plates for feeding and RNAi
This step should be done in the morning. L1-stage synchronized worms are distributed onto each 96-well bacterial clone for feeding and RNAi.
- Retrieve the 96-well RNAi-NGM plates from 37°C and leave them at room temperature to cool down.
- Retrieve the bleached worms from 25°C. Estimate the number of worms per 2 μL and adjust with M9 to have around 100-120 worms per 2 μL (roughly a single drop).
- Distribute manually 2 μL of L1-stage worms in each well using a repetitive dispenser. Check each well for worms (you should see worms swimming in liquid).
- Let the plates dry under a sterile laminar flow cabinet (maximum 1 hour). Check the plates regularly; the worms must be crawling and not swimming. Be careful, the NGM must not dry too much, otherwise it will crack.
- Place the plates at 25°C in a humid chamber.
7. Test the Drechmeria coniospora spores for effective infection
This step is specific for the present screen. It consists in testing different batches of spores to select highly infective ones. Therefore it should be done as close as possible before day 4, the day of infection. It is necessary to prepare sufficient L3-L4 worms in advance for these tests (Table 1). The detailed methods required for D. coniospora culture have been described elsewhere 11.
- Collect spores from a sample of each fungal culture in a small volume of M9.
- Place a drop of each spore suspension on a 35 mm NGM plate seeded with OP50, and let it dry.
- Add 30-40 L3-L4 worms carrying the reporter gene of interest (e.g. pnlp-29::GFP).
- Place the infected plates at 25°C ON.
- Next day, check the worms for GFP induction and select the batch of fungus that gives the brightest, broadest and most homogenous induction of green fluorescence.
Day 4
8. Infect the RNAi exposed worms with D. coniospora spores
This step is performed 30 hours after RNAi feeding when worms have reached the L3-L4 stage.
- Determine the volume of spores to use to distribute 4 μL per well.
- Collect fresh D. coniospora spores from the selected batch with M9 buffer. Use 8-10 mL of M9 for one 9 cm plate of spores.
- Distribute 4 μL of spores to each well with a repetitive dispenser. Check each well for spores (you can see the worms swimming in liquid).
- Let the plates dry under a sterile laminar flow cabinet (~ 1 hour). Check the plates regularly until the worms start crawling. Be careful, the NGM must not dry too much, otherwise it will crack.
- Place the infected plates at 25°C in a humid chamber.
Day 5
9. Observation and storage of the plates before automated quantitative analysis
This step starts 18 hours after infection and is performed during day 5. Worms are expected to be young adults starting to lay eggs. During this step the phenotype is scored visually: either worms fluoresce green which corresponds to a normal situation following infection, or the induction of the GFP is altered in a given well which means that the silenced gene changes the response to infection.
- Rapidly observe the plates under the fluorescence dissecting microscope. If the plates show a good general GFP induction then proceed to the next step, otherwise wait until the afternoon for a better induction, as long as there is still food left for the worms.
- Visually score every plate and note any well with a marked phenotype (e.g. no expression of GFP). By checking for the results obtained with any control RNAi clones that may have been added, this first preliminary analysis gives an indication of whether the experiment has been successful.
- Using a 8 or 12-channel Multi Pipette transfer the worms with 100 μL of NaCl 50 mM-Triton 0.05%, to a new correspondingly labeled/barcoded 96-well round-bottomed plate. Freeze the plates at -80°C until analysis.
- On the day of the analysis, thaw the plates at room temperature and proceed to the automated analysis using the COPAS Biosort. The sorter analyzes a single plate in 22 minutes. Plates should not be left at room temperature for more than an hour before analysis, and they cannot be frozen again.
- The information obtained from the COPAS Biosort are transcribed into an Excel file for cursory verification of data quality. The raw data with the different parameters measured by the COPAS Biosort (optical density (extinction), axial length (time of flight), fluorescence emissions, simultaneously by three different detectors) are then stored in a dedicated LIMS package (MBio LIMS, Modul-Bio) for subsequent detailed quantitative analyses.
10. Representative Results
Using the quantities of worms and bacteria given above, at the end of the experiment, in most wells, the animals should all have developed to adulthood and there should still be some food left in all wells. Under these conditions, most wells should also contain eggs. There should be a sufficient number of adults in each well so that Biosort data is obtained for at least 50 individual worms. On the other hand, if the RNAi is efficient, a large number of RNAi clones will provoke a visible phenotype. For example, worms may be uncoordinated, or more obviously, sterile, or arrested in their development. This should occur frequently, so that generally at least one well per 96-well plate contains worms with an evident RNAi phenotype. When the phenotype of interest is the expression of a GFP reporter gene, wells with a gfp(RNAi) clone provide a robust control (Fig. 2). Other phenotypes, such as an alteration of size can be readily measured with the Biosort (Fig. 3A). We observed that worms could migrate to adjacent wells at very low frequency (<1%, BS, unpublished results) when the food in a well did run out.
Knocking down the function of different classes of genes can affect transgenes expression in C. elegans. Some are non-specifically required for transgenes expression 12. Others, such as tissue-specific transcription factors, for example the epidermal GATA factor ELT-3 13, will be required in particular subsets of cells. This is one of the reasons for the inclusion of a non-related and constitutive epidermal cell transgene reporter construct (pcol-12::dsRed) in the strain used for the current protocol. This allows such genes to be distinguished from those specifically involved in the gene regulatory process under study (Fig. 3B & C). This transgene is expressed during all larval stages. If an assay involves detecting gene expression in embryos a different control reporter gene would be required.
One of the innovations of this protocol is the use of freezing plates to postpone their analysis. Freezing allows plates to be stored for up to two weeks without substantially altering the results. Although the absolute values of measured fluorescence may be changed, their relative ratios remain similar (Fig. 4). On the other hand, RNAi is an intrinsically variable experimental procedure 14. To obtain robust results and eliminate the many potential false positives, RNAi screens need to be replicated, at least once. If the experiments are successfully conducted, there should be a reasonable correlation between the results from one plate tested on different days. Particularly, the genes that have a strong effect should be clearly identifiable, even if their precise quantitative effect is not identical between duplicate plates (Fig. 5).
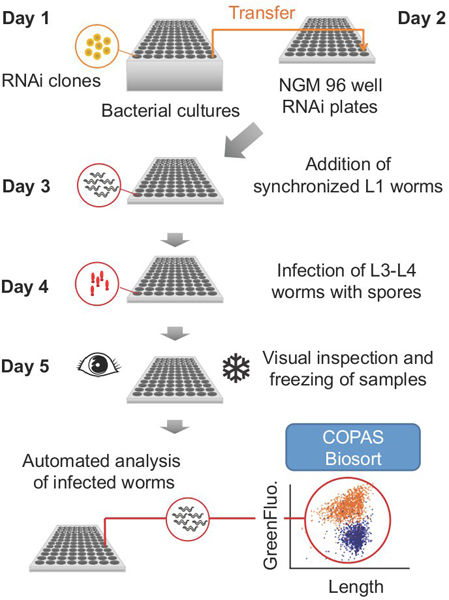
Figure 1. Overall scheme for a typical experiment.
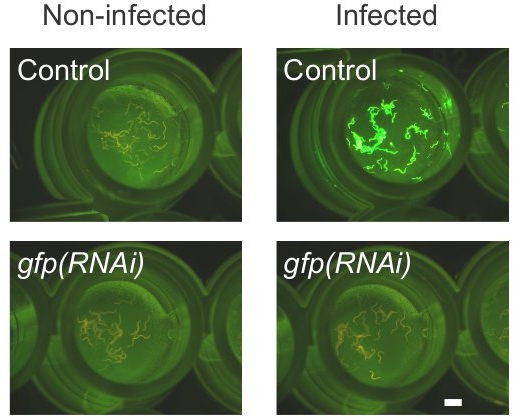
Figure 2. Control RNAi using a clone targeting GFP expression. Transgenic worms expressing constitutively a pcol-12::dsRed reporter and an inducible pnlp 29::GFP reporter on solid medium in a 96-well plate visualized using a GFP dissecting microscope with a filter set allowing simultaneous observation of red and green fluorescence. Worms were exposed to control (upper panels) or a GFP RNAi clone (bottom panels) for 48 h. The panels on the left are uninfected worms, those on the right worms 18 hours after infection with D. coniospora. The scale bar is 1 mm.
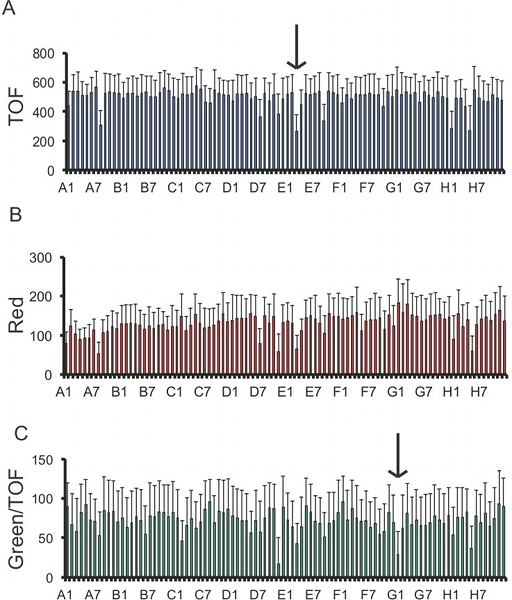
Figure 3. Quantitative analysis of a 96-well plate. The COPAS Biosort generates numerical data for each worm analyzed. The graphs show the average and standard deviation for the time of flight (TOF; A) which corresponds to the size of the worms 15, red fluorescence (B) and the ratio (X100) of green fluorescence to TOF (C) of transgenic worms expressing constitutively a pcol-12::dsRed reporter and an inducible pnlp 29::GFP reporter that had been cultured on solid medium in a 96-well plate with different RNAi clones for 48 h, 18 hours after infection with D. coniospora. Certain clones, such as the one highlighted with the arrow in (A), provoke growth defects and/or affect the expression of pcol-12::dsRed. Others, specifically affect GFP expression, as highlighted by the arrow in (C). This particular clone targets the gene nsy 1, previously shown to be important for the regulation of nlp 29 13. Green, red fluorescence and TOF are measured in arbitrary but constant units.
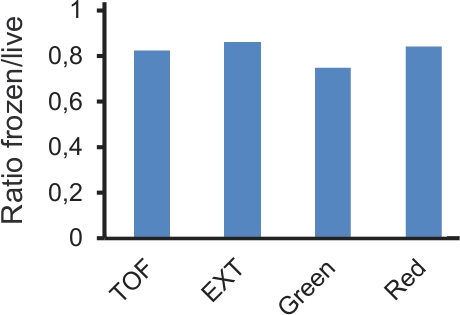
Figure 4. Comparison of data obtained with live and frozen samples from the same experiment. Plates of L4 worms carrying pnlp-29::GFP and pcol-12::dsRed transgenes were either infected with D. coniospora or not. After 18h, each plate was roughly divided in 2; half was analyzed immediately and half frozen at -80°C for 24h, before thawing and analysis. Values for average TOF (size of worms), EXT (optical density) and Green and Red fluorescence were calculated for each sample. The graph shows the ratio of the average for infected divided by non-infected samples, for the frozen and live samples (n= 7638 and 5096, and 8634 and 3850 worms, respectively).
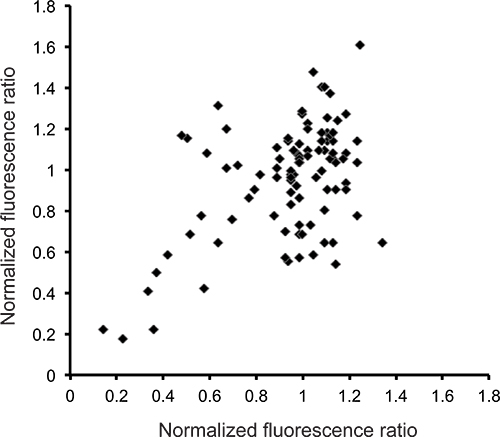
Figure 5. Comparative analysis of duplicate 96-well plates. The normalized fluorescence ratio (the mean fluorescence ratio (here, 100*green/TOF) for each well divided by the trimmed (25th to 75th percentile) mean for each plate) for each well from duplicate analyses of a representative 96-well plate.
| Material | Experimental Timeline |
|
Thursday
|
Table 1. Example of a one-cycle experiment with 30 plates.
Here is presented the material needed for a one-cycle experiment of 30 plates and a timeline for an experiment from step 1 to step 12 organized on 9 days.
Nematode Growth Media (NGM), 100 mL:
| 0.3 g | NaCl |
| 0.25 g | BactoPeptone |
| 2 g | BactoAgar |
| 100 μL | 5 mg/mL Cholesterol in EtOH |
| Add deionized water to 100 mL | |
| Autoclave for 5 minutes at 121°C and let cool, then add: | |
| 100 μL | 1 M MgSO4 |
| 100 μL | 1 M CaCl2 |
| 2.5 mL | 1 M KPO4 pH 6.0 |
NGM for RNAi treatment in 96-well plate, 100 mL:
| 0.29 g | NaCl |
| 0.25 g | BactoPeptone |
| 1.7 g | BactoAgar |
| 100 μL | 5 mg/mL Cholesterol in EtOH |
| Add deionized water to 100 mL | |
| Autoclave for 5 minutes at 121 °C and let cool, then add: | |
| 100 μL | 1 M MgSO4 |
| 100 μL | 1 M CaCl2 |
| 2.5 mL | 1 M KPO4 pH 6.0 |
| 400 μL | 1 M IPTG |
| 100 μL | 100 mg/mL Ampicillin |
| 100 μL | 12.5 mg/mL Tetracycline |
Bleach solution, 5mL:
2.5 mL H2O
2.3 mL Bleach
0.2 mL NaOH 50%
NaOH 50%, 100 mL:
50 g NaOH
Add deionized water to 100 mL
NaCl 50mM-Triton 0.05%, 400 mL:
4 mL NaCl 5 M
1 mL Triton 20%
Add deionized water to 400 mL
Triton 20%, 10 mL:
2 mL Triton X-100
8 mL deionized water
M9 buffer, 1L
6 g Na2HPO4 (MW : 178)
3 g KH2PO4 (MW : 136)
5 g NaCl
Add deionized water to 1 L
Autoclave
Add 1 mL MgSO4 1 M
Luria Broth (LB), 1L:
10 g BactoTryptone
20 g Yeast extract
10 g NaCl
Add deionized water to 1 L
Autoclave
1 M MgSO4, 300 mL:
73.95 g MgSO4
Add deionized water to 300 mL
Autoclave and store at room temperature
5 mg/mL Cholesterol, 200 mL:
1 g Cholesterol
Add 100% EtOH to 200 mL
Filter sterilize and store at room temperature
1 M CaCl2, 500 mL:
27.75 g CaCl2
Add deionized water to 500 mL
Filter sterilize and store at room temperature
1 M KPO4 pH 6.0, 4 L:
517 g KH2PO4 (MW : 136)
207 g K2HPO4 (MW : 174)
Add deionized water to 4 L
100 mg/mL Ampicillin (Amp), 10 mL:
1 g Ampicillin
Add deionized water to 10 mL
Store at -20 °C
1 M Isopropyl β-d-Thiogalactopyranoside (IPTG), 10 mL:
2.38 g IPTG
Add deionized water to 10 mL
Store at -20 °C
12.5 mg/mL Tetracycline, 100 mL
1.25 g Tetracycline
Add 100% EtOH to 100 mL
Table 2. Solutions.
