The level of expression achieved for trpLE fusions is variable and heavily dependent on the amino-acid sequence of the attached peptide. Figure 3 shows the SDS-PAGE analysis of pre-induction (lane 1) and time-of-harvest (lane 2) samples from a culture that yielded approximately 120 mg of pure, intact trpLE-DAP12TM fusion from 1 liter of culture and 4 ml nickel matrix. All of the trpLE-DAP12TM fusion was localized to the inclusion body pellet (lane 4) as opposed to supernatant (lane 3).
Approximately 70% of the nickel-purified trpLE-DAP12TM fusion was disulfide-crosslinked to the dimeric form (Figure 3, lanes 5 and 6: non-reduced and reduced samples of TFA eluate). This is a typical result for the trpLE-DAP12TM fusion, but crosslinking efficiency will vary with the placement of engineered cysteines. The DAP12TM peptide products are not readily identifiable in total CNBr digest samples (lanes 7 and 8), but comparison of the intact fusion and trpLE fragment bands in the reduced digest sample (lane 8) indicates that digestion of the fusion was ~60% complete.
Figure 4 shows the separation of digest products by reversed-phase HPLC using a preparative-scale C3 column (total binding capacity ~200 mg of protein). Because the aromatic content of the fusion is low we typically monitor the absorbance at 214 nm rather than 280 nm. For this run 90 mg of oxidized, CNBr-digested trpLE-DAP12TM fusion was dissolved in 3 ml neat formic acid and loaded onto the column in 100% solvent A (60% H2O, 40% acetonitrile, 0.1% TFA). Bound products were eluted in a two-stage gradient of 0-60% followed by 60-90% solvent B (75% isopropanol, 25% acetonitrile, 0.1% TFA) at a flow rate of 10 ml/min. The major products contained in each fraction according to SDS-PAGE analysis (Figure 5) are indicated above the chromatogram using the codes assigned in Figure 2. See Optimization of HPLC gradient elution in the DISCUSSION section for more details on optimizing gradient conditions for different peptide sequences.
Peptide products are easily identifiable in the SDS-PAGE (Figure 5) and MALDI-TOF (Figure 6) analyses of the major HPLC fractions. DAP12TM peptides and, in our experience, many other hydrophobic TM peptides, run with significantly reduced migration rates compared to their expected molecular weights (3366 Dalton monomer and 6730 Dalton dimer for the 15N-labelled species shown here). This behavior is directly related to the quantity of peptide loaded on the gel and results in a continuum of apparent molecular weights as the amount loaded is varied (data not shown). However, the alteration in electrophoretic mobility of peak 5 upon reduction with 100 mM DTT (compare lanes 7 and 8) identifies this as the crosslinked peptide product. MALDI-TOF MS analysis confirms the identity of the dimeric peptide with the major peak at 6729.2 Daltons and reveals no other major products (Figure 6). The final yield of pure disulfide-crosslinked DAP12TM peptide homodimer from this preparation was 7.5 mg per liter of culture.

Figure 1. DNA sequence of the trpLE-DAP12TM coding region with its amino acid translation. Key regions of the polypeptide sequence are highlighted and color-coded with labels to the right. The unique internal methionine (Met) and cysteine (Cys) residues for cyanogen bromide (CNBr) cleavage and oxidative crosslinking are highlighted in cyan and purple, respectively. HindIII and BamHI restriction sites (bold and underlined) are unique in the plasmid and may be used to replace the TM peptide sequence with another sequence of interest.
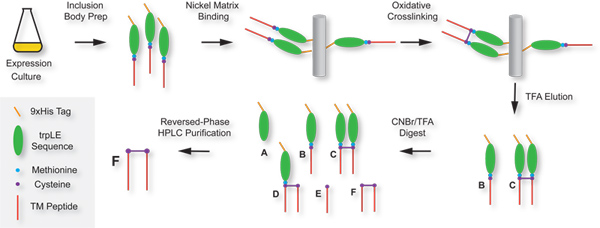
Figure 2. Schematic diagram of the procedure. The major steps in the protocol are indicated in text labels. Key segments of the trpLE-DAP12TM fusion are color-coded as in Figure 1. Major polypeptide products from the crosslinking and digest steps are labeled A-F and are listed here with their description and expected molecular weights (15N-labeled): [A] trpLE fragment: 13,769 Daltons; [B] intact trpLE-DAP12TM fusion: 17,118 Daltons; [C] intact trpLE-DAP12TM fusion dimer: 34,233 Daltons; [D] single-cleaved trpLE-DAP12TM fusion dimer: 20,482 Daltons; [E] monomeric DAP12TM peptide: 3366 Daltons; [F] dimeric DAP12TM peptide: 6730 Daltons. Click here to view larger figure.
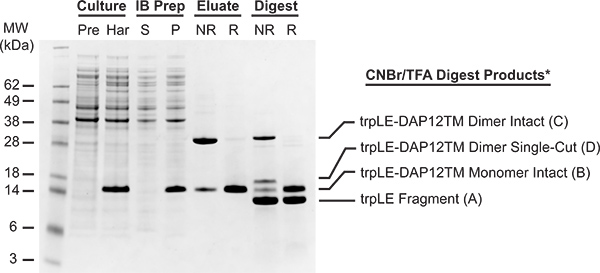
Figure 3. SDS-PAGE analysis of trpLE-DAP12TM expression, purification, crosslinking and digest steps. Culture samples (lane 1, pre-induction, step 2.5; lane 2, harvest, step 3.1) and inclusion body prep samples (lane 3, supernatant; lane 4, pellet; step 3.4) were reduced with 100 mM DTT. TFA eluate from nickel column (lanes 5 and 6; step 6.1) and post-CNBr sample (lanes 7 and 8; step 6.3) were run non-reduced (NR) and reduced with 100 mM DTT (R) as indicated to confirm the disulfide-crosslinked products. *Monomeric and dimeric peptide products E and F are poorly visualized by Coomassie staining and therefore are not detectable on this gel.
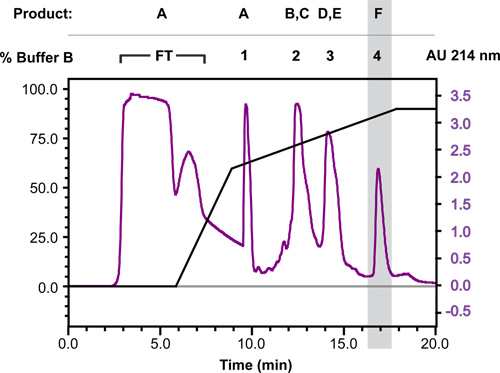
Figure 4. Reversed-phase HPLC purification of DAP12TM disulfide-crosslinked dimer. Oxidized, CNBr-digested and lyophilized trpLE-DAP12TM fusion products (90 mg) were dissolved in 3 ml formic acid (100%) and loaded onto an Agilent ZORBAX SB-300 C3 PrepHT (21.2 x 150 mm) column in solvent A (60% H2O, 40% acetonitrile, 0.1% TFA). A two-stage gradient of 0-60% followed by 60-90% solvent B (75% isopropanol, 25% acetonitrile, 0.1% TFA) was run at a flow rate of 10 ml/min to elute bound products. Fractions collected as flow-through (FT) and four major peaks are marked above the 214 nm absorbance trace (AU: arbitrary units) and the major products contained in each fraction are indicated using the codes assigned in Figure 2. The desired DAP12TM crosslinked product peak is shaded.
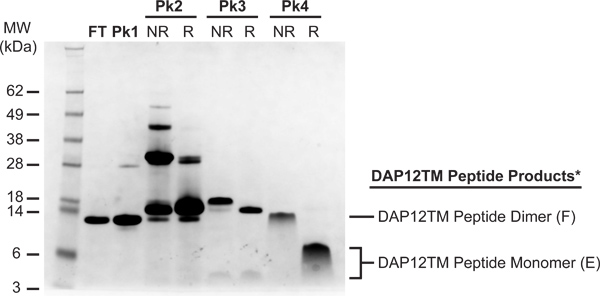
Figure 5. SDS-PAGE analysis of reversed-phase HPLC products. Flow-through (FT) and peak 1 samples were run non-reduced. Peak 2, 3 and 4 samples were run both non-reduced (NR) and reduced by treatment with 100 mM DTT (R) to verify the identity of disulfide crosslinked products. The major products in each fraction (with their codes assigned in Figure 2) were [FT, Pk1]: A, trpLE fragment; [Pk2]: B, intact trpLE-DAP12TM fusion monomer and C, intact fusion dimer; [Pk3]: D, single-cleaved trpLE-DAP12TM dimer and E, monomeric DAP12TM peptide; [Pk4]: F, disulfide-crosslinked DAP12TM dimer. *As discussed (see REPRESENTATIVE REULTS), a concentration-dependent shift in mobility makes the low-MW products in Pk3 (NR and R) and Pk4 (R) appear to be different molecular weights even though both products are monomeric peptide.
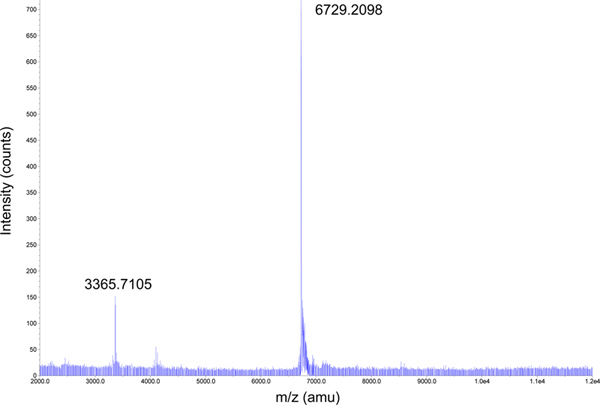
Figure 6. MALDI-TOF mass spectrometric analysis of HPLC-purified DAP12TM peptide dimer. A sample of HPLC fraction 4 (1 μl) was mixed 1:1 with a saturated solution of sinapinic acid (SA) matrix in acetonitrile and spotted on top of a thin layer of dried SA matrix from a saturated solution prepared in ethanol. MALDI-TOF analysis reveals a product at the expected DAP12TM peptide crosslinked dimer mass of 6730 Daltons (6729.2098; 15N-labeled) as well as a minor product at 3365.7105, most likely the double-ionized species. Click here to view larger figure.
