Results are shown in Figure 3 for application of these colorimetric assays to known reference compounds. Representative qualitative data are shown for the Benedict’s (a), Bial’s orcinol (b), and Dische’s diphenylamine (c) assays, and standard curves for these three assays are shown in Figure 4. In panels 3(a-c), the left panels show positive/negative control experiments using suitably reactive/unreactive analytes; these visual results are shown in situ, in the cuvettes as described in Protocols 1-3 (above). The right sub-panels show a titration series for each respective analyte. In the Benedict’s assay (a), the positive control is ribose (0.42 mg/ml), while negative controls are water, a generic protein (0.75 mg/ml BSA), and two non-reducing sugars (DNA at 0.75 mg/ml and RNA at 12.5 mg/ml). In the orcinol assay (b), the positive controls are ribose (0.15 mg/ml), RNA (7.5 mg/ml), and DNA (0.45 mg/ml), while water and BSA protein (0.45 mg/ml) are negative controls. In the diphenylamine assay (c), calf thymus DNA (0.45 mg/ml) is shown as a positive control, and water, yeast extract RNA (7.5 mg/ml), ribose (0.15 mg/ml), and BSA (0.45 mg/ml) serve as negative controls. Finally, panel (d) illustrates the robustness of the assays by showing the Dische’s reaction with samples of varying heterogeneity: two concentrations of DNA are shown as a positive control in the left two samples (cuvettes 1-2), DNA + RNA mixtures (at various ratios) are shown in the next three samples (3-5), and the final three samples show DNA in the absence (sample 6) or presence (samples 7-8) of the nucleic acid-binding protein ‘Hfq’.16 Note that the positive result of the Dische’s assay is preserved for DNA-containing samples even in the presence of ‘contaminating’ RNA or protein.
Quantitative Analysis: The dilution ranges in the Figure 3(a-c) panels vary because each reaction has a distinct visual detection limit, depending on the type of sugar being assayed. Spectrophotometric, rather than visual, detection can be used to improve the measurement ranges, e.g. as shown in Figure 4 for the (a) Benedict’s, (b) Bial’s, and (c) Dische’s assays. Though the protocols described here are intended as primarily qualitative assays, the Beer-Lambert relation between absorbance and concentration enables at least semi-quantitative estimation of the sugar or nucleic acid content. As an example of a standard curve for calibration of the Benedict’s test, a linear regression fit of absorbance (475 nm) against ribose concentration is shown in Figure 4(a); error bars indicate the standard deviation of n = 3 replicates, and the squared correlation coefficient is provided. The deviation from linearity at very low ribose concentrations (e.g. the 0.28 mM datum) reflects the limited sensitivity of this assay. Though the assays are primarily intended for qualitative studies, the following guidelines are suggested for semi-quantitative analysis:
- For the Benedict’s assay (Figure 4a): The reaction readout (A475nm) was found to be linear at least over the range 0.04 → 0.5 mg/ml analyte, as performed in triplicate.
- For the Bial’s assay (Figure 4b): The absorbance data (A660nm) were linear at least over the range 0 → 0.5 mg/ml analyte (baker’s yeast RNA), the assay having been performed in triplicate (R2 = 0.99 linear regression coefficient). Though the samples were centrifuged for clarification before absorbance measurements, no precipitant was found at these low concentrations of analyte and therefore the centrifugation step was not strictly necessary. The assay deviates from linearity at analyte concentrations beyond this range.
- For Dische’s assay (Figure 4c): The A600nm response was found to be linear at least over the range 0.15 → 0.75 mg/ml of analyte (calf thymus DNA), the assay having been performed in triplicate (R2 = 0.99). The plot of absorbance versus [analyte] began to plateau at 0.90 mg/ml DNA, indicating the boundary of the linear response region.
- Practical considerations for quantitative or semi-quantitative analysis: To remove precipitated material that would otherwise interfere with absorbance readings, all three assays require centrifugation at maximal speeds (25,000 x g, or whatever limit can be withstood by the microfuge tubes) for reasonable lengths of time (e.g., 20 min); this is especially important at higher analyte concentrations. After centrifugation, clarified samples can be transferred to a cuvette or microplate (e.g., 96-well microtiter plates) for convenient absorbance measurements.
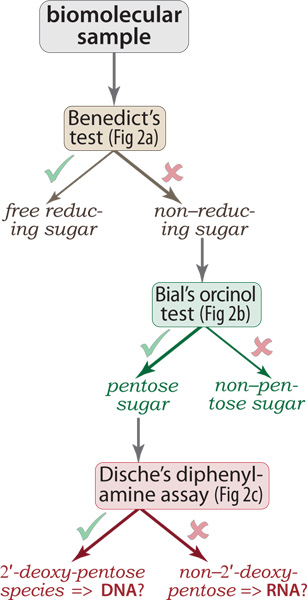
Figure 1. Decision tree for application of the assays. Starting with a potentially heterogeneous sample of biomolecules (e.g., from whole-cell lysates), the colorimetric assays described here can be used to determine if the mixture contains non-reducing sugars (Benedict’s test). If so, then Bial’s orcinol test further reveals whether the population of non-reducing sugars contains pentose rings (as in DNA or RNA), versus hexoses (e.g., glucose, other pyranoses) and possibly yet other aldoses. Finally, if the sample contains at least a moderate fraction of DNA then the 2′-deoxyribose ring will react (upon acidification and heating) with Dische’s diphenylamine reagent, yielding a visibly blue condensation product. Note that this diagram is a decision tree, not a flowchart: the logic of the assay results is shown serially, but the assays can be executed in parallel.
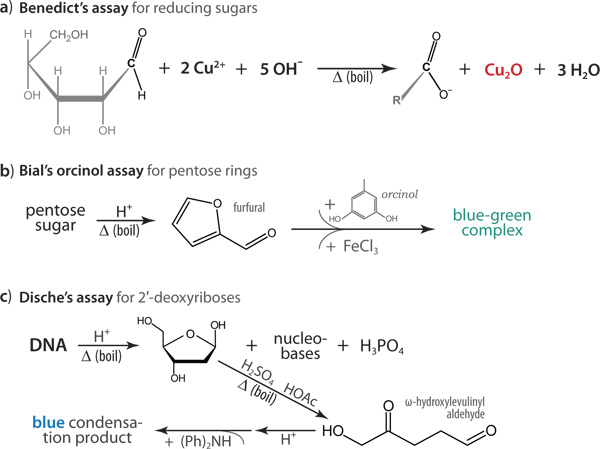
Figure 2. Underlying chemical reactions are shown for the colorimetric assays, with the detectable colored product indicated alongside the corresponding reactions. (a) An insoluble, red precipitate of Cu2O is the positive result of Benedict’s assay for reducing sugars. (b) Upon heating and acidification, sugars containing pentose rings will decompose to furfural, which then reacts with orcinol (Bial’s reagent) to yield a soluble blue-green adduct. In Dische’s assay, lack of a hydroxyl substituent at the 2′ position enables a ring-opening oxidation reaction, the aldehyde product of which further reacts with diphenylamine [(Ph)2NH] to yield a bright blue product. Chemical structures remain unknown for the large, multi-ring condensation products of the orcinol (b) and diphenylamine (c) reactions.

Figure 3. Application of the colorimetric assays to reference compounds and heterogeneous samples. Sample results are shown for the Benedict’s (a), Bial’s (b), and Dische’s (c) colorimetric assays. In (a) → (c), the left sub-panels show positive/negative controls using suitably reactive/unreactive analytes and the right sub-panels show a titration series for each analyte. Also shown is an illustrative panel of Dische reactions (d) wherein the analyte varies in heterogeneity – either DNA alone (left), DNA/RNA mixtures of differing ratios (middle), or DNA in the presence of a nucleic acid-associated protein (‘Hfq’). These panels are further described in the Representative Results section of the text.
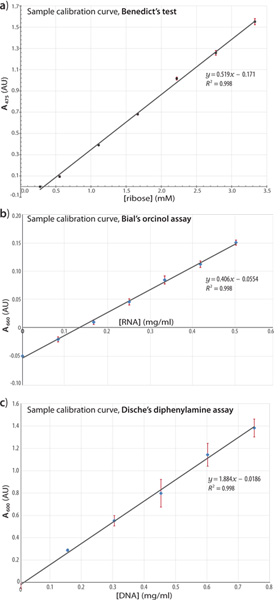
Figure 4. Standard curves from assays with reference compounds. Calibration curves are shown for the Benedict’s (a), Bial’s (b), and Dische’s (c) assays, depicting a representative portion of the linear response region for each assay. The linear regression fits and corresponding correlation coefficients are indicated for each assay. Standard baker’s yeast RNA (b) and calf thymus DNA (c) were the analytes in these titration series. Click here to view larger figure.
