To characterize the dynamic behavior of the virus particles, HeLa cells infected with HIV-1 were imaged by high-speed confocal live-cell microscopy and the particle movements were analyzed by automated 3D particle tracking (Figure 1). To avoid the time lapse of several minutes that can occur between collection of the last confocal live-cell image and plunge-freezing (which may be long enough to lose correlated HIV-1 particles), a cryo-fluorescence light microscopy stage (Figure 3) was designed and constructed for imaging frozen-hydrated samples15, within cryoEM cartridges, on light microscopes. A copper block platform (Figure 3b, black arrow) and copper ring (Figure 3d, arrowhead) was added for use with non-Polara electron microscopes. Correlation between the confocal live-cell, cryo-fluorescence light microscopy, and cryoET was achieved using indexed gold Quantifoil grids and 0.2 μm fluorescent latex beads (Figure 2). Taken together, Figure 4 demonstrates the feasibility of the overall procedure for advanced correlative live-cell microscopy and cryo-electron tomography to visualize fluorescently labeled HIV-1 particles interacting with a host HeLa cell.
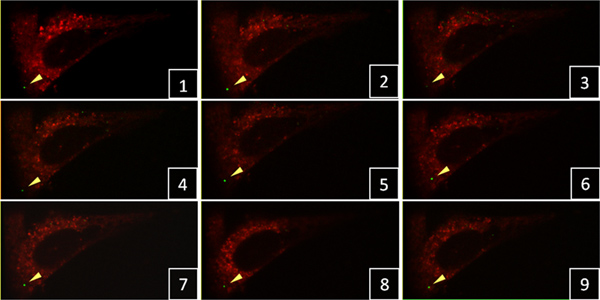
Figure 1. Time-lapse confocal live-cell imaging of HIV-1 particles in HeLa cells. A single green-fluorescent viral particle (yellow arrowhead) was tracked in 3D confocal stacks with a 3 min time interval between frames. Click here to view larger figure.

Figure 2. Correlation between fluorescence images and cryoEM images. (a & b) Differential interference contrast (DIC) images of HeLa cells cultured on a Quantifoil gold EM finder grid, recorded with a 2X (a) and 20X (b) objectives. (c) Cryo-fluorescence image of frozen-hydrated HeLa cells (red color for mitochondria) and 0.2 μm fluorescent latex beads (green), recorded with a cryo-stage and a 40X long working distance air objective, overlaid with a DIC image of cells. (d) Low magnification cryoEM image of the corresponding region in (c). (e & f) Correlation between fluorescence and cryoEM images using 0.2 μm fluorescent latex beads. Corresponding beads are circled with the same color. Scale bars, 200 μm in a, 50 μm in b, 25 μm in c, d & e, 5 μm in f.

Figure 3. Construction of cryo-light microscopy stage. (a) A self-pressurized dewar, filled with liquid nitrogen, used to cool down the copper cryo-sample stage (white arrow). (b) Top, inside view of the cryo-sample stage, showing the inner chamber (white arrowhead). The sample grid is placed onto the EM specimen cartridge, which sits in the center of a copper block (black arrow). Once loaded with the sample grid, the cartridge is transferred to the inner chamber (white arrowhead) for cryo-fluorescence light microscopy. (c & d) The specimen cartridge before (c) and after (d) placing a copper ring (black arrowhead) to keep the grid in place for using with non-Polara cryo-electron microscope.

Figure 4. CryoET of a single fluorescent particle bound to the cellular protrusion of a HeLa cell by correlative microscopy. (a & b) A DIC image recorded with a cryo-light microscopy stage (a) overlaid with a cryo-fluorescence image of GFP-tagged particles (red) (b). (c – e) Low dose cryoEM images of the region containing a fluorescent particle (circled in panel b & c) at 140X (c), 3,500X (d) and 27,500X (e), respectively. Inset, an enlarged view recorded after acquisition of the tomographic tilt series. (f – h) Three 4 nm thick tomographic slices separated by a distance of 21 nm in the z direction. Connections between the particle and HeLa cell membrane are indicated by arrows in both the projection image (inset in panel e) and tomographic slices (panels f & g). Scale bars, 10 μm in a & b, 20 μm in c, 500 nm in d, and 100 nm in e – h.
