Fluorescent labeling of cytoplasmic InlC provides a robust readout for cell infection by L. monocytogenes, as illustrated in Figure 1: the central cell in the micrograph is highly infected by the strain P14.PrfA*23 as it can be observed in the phase contrast image (arrowheads, Figure 1A) and it is confirmed by the DAPI signal where individual bacteria can be clearly distinguished (Figure 1B). The InlC staining (superposed to the DAPI staining in Figure 1C) depicts how this secreted protein densely accumulates in the cytosol of infected cells and allows an unambiguous detection of the infected host cell morphology. Staining of the actin cytoskeleton with fluorescent phalloidin (Figure 1D) provides information concerning the morphology of the complete inoculated cellular monolayer and further illustrates how neighboring non-infected cells (cell nuclei marked with an asterisk) do not display any InlC labeling. It is worth to mention that, since InlC cytosolic levels are dependent on the number of cytoplasmic bacteria, we have noted a correlation between the InlC signal detected by immunofluorescence and the number of L. monocytogenes per cell: as observed in Figure 1, neighboring cells infected with low bacterial numbers display a reduced InlC staining which can be quantified. Interestingly, it is also possible to observe that InlC is easily detected in the protrusions formed by bacteria caught in the process of cell-to-cell spread (Figure 1C) as observed also with the actin staining (Figure 1D). Of note, bacteria are not directly stained in our protocol but since the 488 nm channel is not used, L. monocytogenes can be labeled using a specific anti-bacterial serum; otherwise, GFP-expressing bacteria can alternatively be used.
Using imaging analysis tools such as the public software CellProfiler (Broad Institute, www.cellprofiler.org), it is possible to segment cells and to measure the InlC signal in individual infected cells, which can be used to estimate an infection index for a specific experimental condition. As shown in Figure 2, the labeling of the nuclei with DAPI (Figure 2A) and the actin cytoskeleton with phalloidin (Figure 2C) provide the information required for cellular segmentation: the nuclei are used as reference objects for the identification of individual cells (Figure 2B) and the cytoplasm is subsequently identified using a spread function from the nuclei that takes into account the cytoskeletal signal (Figure 2D). Finally, the intensity of the InlC signal can be quantified for each identified cellular object (Figures 2E and 2F) to estimate the number of infected cells by setting a threshold for the InlC intensity that we consider as negative. The infection index is calculated as the number of infected cells divided by the total number of cells in the population.
Our protocol can be coupled to siRNA screens to investigate the function of large panels of target molecules in the infection of host cells by L. monocytogenes. Controls are required to validate the efficiency of transfection: for example, siRNA targeting Kif11 is a commonly used control for transfection that leads to cell death, and therefore the absence of cells at the end of the assay is an indication that transfection proceeded efficiently. To specifically address the L. monocytogenes invasion process, siRNA inactivation of the cellular receptor Met in HeLa cells leads to a major inhibition of bacterial entry into host cells and leads to very low levels of InlC-positive cells in an inoculated cellular monolayer (Figure 3A) as compared to cells treated with a scrambled siRNA control (Figure 3B). The function of candidate unknown molecules can be investigated with our assay using this known cell molecule as standard.
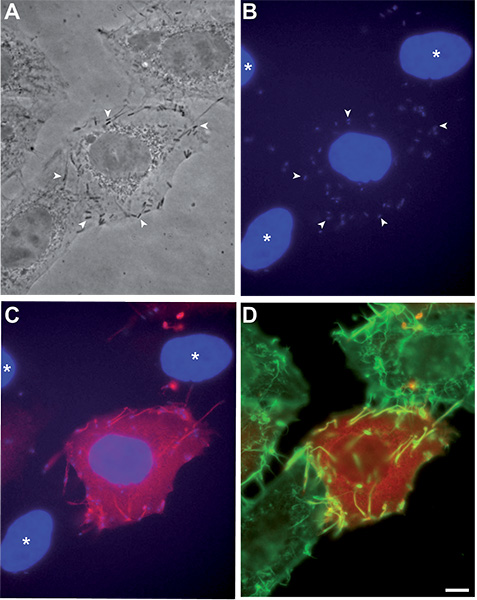
Figure 1. Detection of InlC labeling in HeLa cells infected with L. monocytogenes strain P14.PrfA*. HeLa CCL2 cells have been infected as described in our protocol, processed for immunofluorescence and imaged using a 63X objective. (A) Phase contrast image, several P14.PrfA* bacteria are indicated by arrowheads. (B) DAPI staining (blue), the same individual bacteria as in (A) are labeled by arrowheads. (C) Superposition of the DAPI staining (blue) and the InlC staining (red), the nuclei of uninfected cells are labeled by an asterisk. (D) Superposition of the InlC (red) and actin (green) signals. Bar: 5 μm.
Click here to view larger figure.

Figure 2. Segmentation analysis of HeLa cells infected with L. monocytogenes strain EGDe.PrfA*. HeLa CCL2 cells were infected as described in our protocol, processed for immunofluorescence and imaged using a 10X objective. (A) DAPI signal. (B) Superposition of the DAPI signal (blue) displayed in (A) and actin signal (red) displayed in (C) showing the segmentation of the nuclei (view enlarged inset). (C) Actin signal. (D) Same image as (B) showing the segmentation of the cells. (E) InlC signal. (F) Same image as (D) with superposition of the InlC staining (yellow). Bar: 50 μm.
Click here to view larger figure.
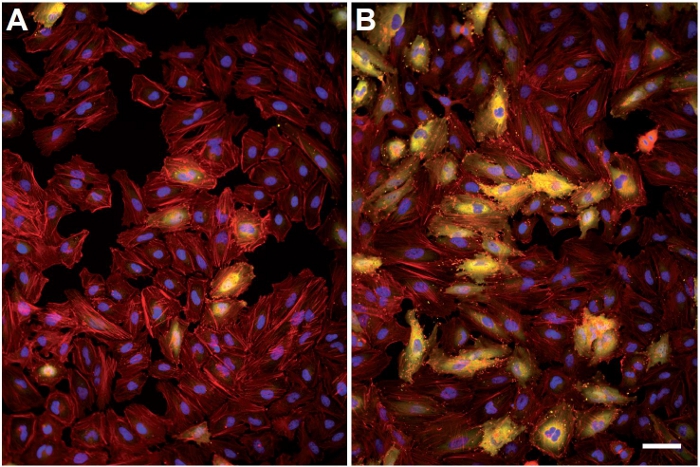
Figure 3. Variation of the InlC labeling between cells inactivated for Met and control cells. HeLa CCL2 cells were initially reverse-transfected with a pool of four siRNAs targeting the host cell receptor Met; 72 hr after transfection, cells were infected as described, processed for immunofluorescence and imaged using a 10x objective. (A) Superposition of the DAPI (blue), actin (red) and InlC (yellow) in cells inactivated for Met. (B) Same channels as in (A) concerning cells treated with a scrambled siRNA control. Bar: 50 μm. Click here to view larger figure.
