HA molecules from A/Shanghai/1/13 and A/Anhui/1/13 expressed well with yields of about 20 mg/L culture. NA molecules of both strains showed moderate expression levels ranging from 0.2 mg/L culture for A/Shanghai/1/13 to 0.7 mg/L for A/Anhui/1/13. All four protein preparations had very little to no impurities (Figures 8A and B) with HAs being of higher purity than NAs which can be explained by the expression level. A/Shanghai/1/13 HA was further analyzed using ELISA with the broadly neutralizing stalk-reactive human mAb CR911423. This mAb binds to a sensitive conformational epitope in the stalk domain and is used here as a probe for correct folding of this domain. The antibody shows good binding which gives confidence that the HA is indeed folded correctly (Figure 8C). A second ELISA with anti-H7 mouse polyclonal serum was performed to confirm the identity of the obtained protein as of H7 origin (Figure 8D).
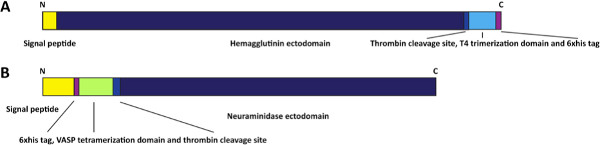
Figure 1. Schematic of HA and NA expression constructs. The expression construct for the HA (A) contains an N-terminal signal peptide followed by the HA ectodomain, a thrombin cleavage site, a T4 trimerization domain and a hexahistidine tag. The NA expression construct (B) contains an N-terminal signal peptide followed by a hexahistidine tag, a vasodilator stimulating phosphoprotein (VASP) tetramerization domain and the NA ectodomain. Click here to view larger figure.
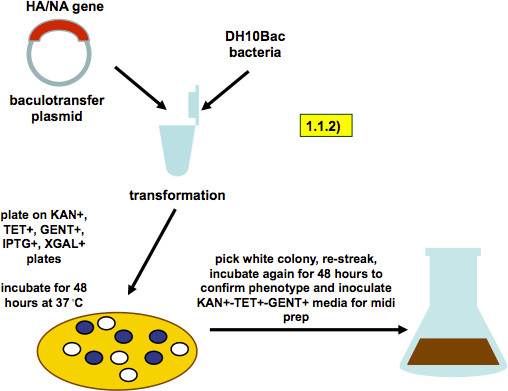
Figure 2. Scheme of bacmid generation (as described in step 1.1.2). A baculovirus-transfer vector containing HA or NA genes is transformed into DH10Bac bacteria. Recombination into the baculovirus genome that these bacteria carry produces a white colony phenotype on selection plates. A white colony is picked and restreaked. A single colony from the restreaked plate is used to inoculate a midiprep culture.
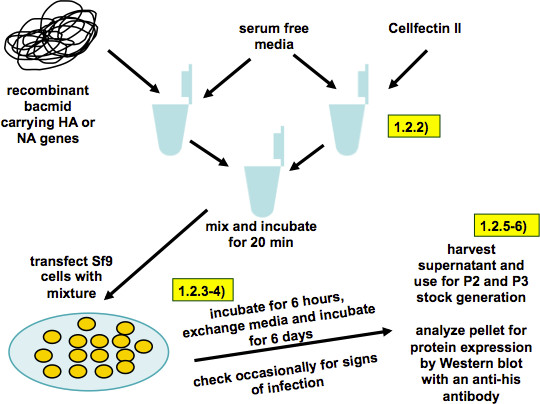
Figure 3. Scheme of baculovirus rescue and working stock generation (as described in steps 1.2.1-6). Recombinant bacmid and transfection reagent are diluted in serum free TNM-FH medium. The two solutions are combined, the mix is incubated for 20 min and then transferred onto Sf9 cells. Six hours post transfection the transfection media is replaced by full TNM-FH media. Six days post transfection the supernatant is harvested and the cell pellet is analyzed for expression of recombinant proteins.

Figure 4. Healthy and infected Sf9 cells. Sf9 cells in (A) are healthy and uninfected. They are attached to the culture dish, look partially polymorphic and relatively small. Cells in (B) are infected with baculovirus, detached from the culture dish, are big and round and have enlarged nuclei. The nucleus also shows a granular appearance.
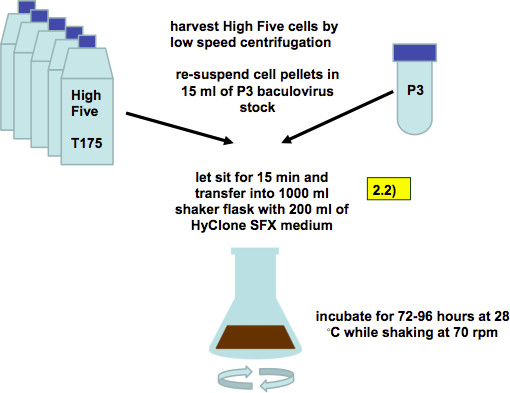
Figure 5. Infection of High Five cells with recombinant baculovirus (working stock, as described in step 2.2). Five confluent T175 flasks with High Five cells are harvested by low-speed centrifugation and combined with 15 ml of P3 baculovirus stock. After 15 min of incubation the cell/virus mixture is transferred into 1,000 ml shaker flasks with 200 ml of SFX media.
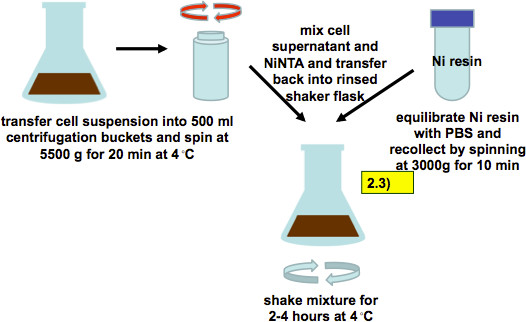
Figure 6. Harvest of cell supernatant 72-96 hr post infection and incubation with Ni-NTA resin (as described in step 2.3). Infected cell suspensions are harvested by low speed centrifugation. Culture supernatants are mixed with PBS-equilibrated Ni-resin and incubated for 2-4 hr while shaking.
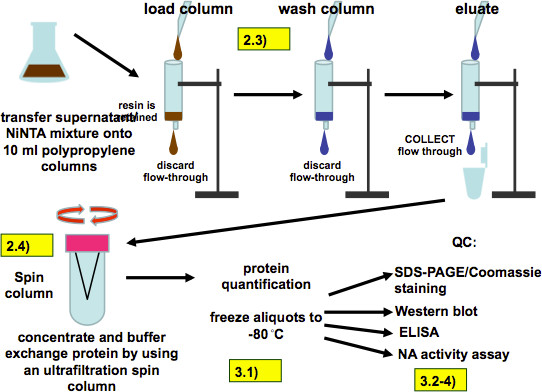
Figure 7. Purification procedure (as described in 2.3 and 2.4) and quality control (as described in steps 3.1.1-4). After incubation the all of the supernatant/resin mixture is loaded on polypropylene columns. The resin is retained by the columns and is then washed four times with washing buffer and the recombinant protein is eluted with 8 ml of elution buffer applied in 2 ml steps. The eluate is then buffer exchanged and concentrated with an ultrafiltration spin column, aliquoted and frozen. Quality control assays include SDS-PAGE, Western Blot, ELISA and measurement of the NA activity of recombinant NA.
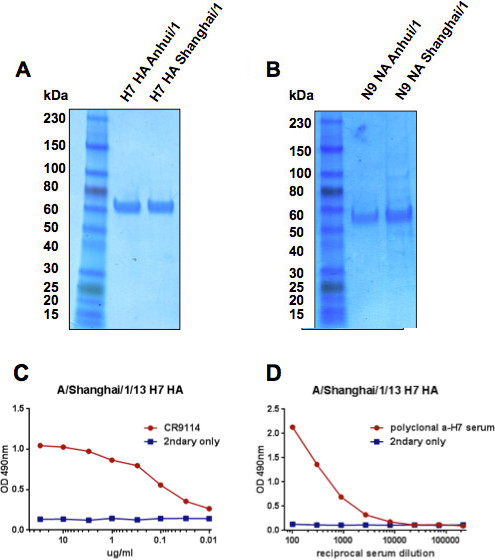
Figure 8. Representative results. HA (A) and NA (B) proteins of A/Anhui/1/13 and A/Shanghai/1/13 were analyzed using SDS-PAGE and Coomassie staining. Structural integrity of the HA of A/Shanghai/1/13 was assessed using the broadly neutralizing human stalk-reactive antibody CR9114 that binds to a sensitive conformational epitope (C). The identity of the same HA was confirmed by a polyclonal anti-H7 mouse serum (D).
