Typical time-course experiments are represented in Figure 2. In these experiments, FRET glutamine sensors with affinities of 8 mM (FLIPQTV3.0_8m, Figure 2A and 2B) and 100 μM (FlipQTV3.0_100 μ C and D) were co-expressed with an obligatory amino acid exchanger ASCT2 18 in cos7 cells 10. Influx of glutamine is detected as the change in fluorescence intensity ratios between the donor (mTFP1) and the acceptor (venus) (Figure 2A and 2C). Efflux of glutamine in the presence of another substrate (Ala) is also clearly demonstrated in these experiments. With both sensors, normalized fluorescent intensities change reciprocally (i.e., the donor intensity goes up as the acceptor intensity goes down, Figure 2B and 2D) in the presence of substrate, which suggest that the ratio change observed are indeed due to the change in FRET efficiency. These experiments show that the glutamine concentrations in these cells fluctuate very dynamically under the experimental condition; from the detection range of 100 μM sensor to the near-saturation concentration for 8 mM sensor.
Substrate specificities of transporters can also be examined using sensors, demonstrated in Figure 3. In these experiments the cells were pre-loaded with glutamine, and then various amino acids were added to the extracellular perfusate to examine whether those amino acids can induce glutamine efflux. As expected, ASCT2 substrates (Ala, Ser, Cys, Thr, D-serine) induced glutamine efflux (Fig. 3A), whereas non-substrate amino acids (Pro, His, Lys) did not (Fig. 3B), corroborating with previous studies 18. Usually, substrate specificity is measured by competition assays using a radio isotope-labeled substrate mixed with competing substrate, which is fairly laborious and requires a population of cells that are evenly expressing the transporter to be studied. Optical imaging exemplified here offers an alternative approach.
When no FRET efficiency changes are observed upon addition of the substrate, several reasons could be considered. One possible reason is the low uptake capacity for the substrate being tested and/or high activities of enzymes that maintain the concentration in the cells. For example, glutamine concentration change was minimal in cos7 cells that do not express ASCT2 transporter under the condition tested, even though this cell line can clearly grow in the media in which glutamine in the main nitrogen source (Figure 4). In addition, if the affinity of the sensor is either too high or too low compared to the intracellular concentration, most of the sensor proteins will remain constitutively bound or unbound respectively; hence no FRET efficiency change will be detected.
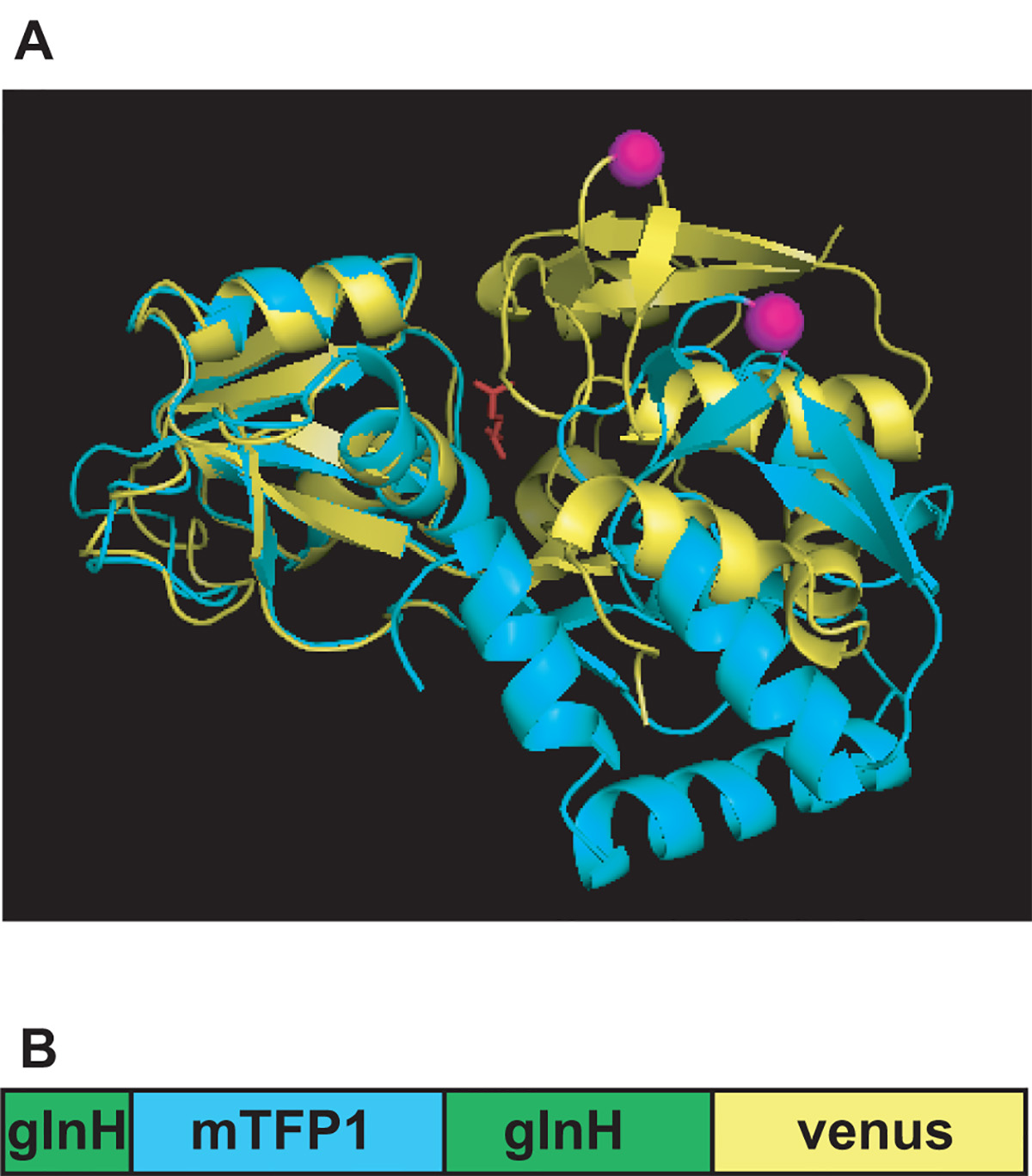
Figure 1. Configuration of a FRET glutamine sensor. (A) Open (cyan) and closed (yellow) conformation of glnH, glutamine binding protein from E.coli. The position where mTFP1 is inserted is marked in magenta. (B) Schematic representations of FLIPQTV_3.0 sensors.
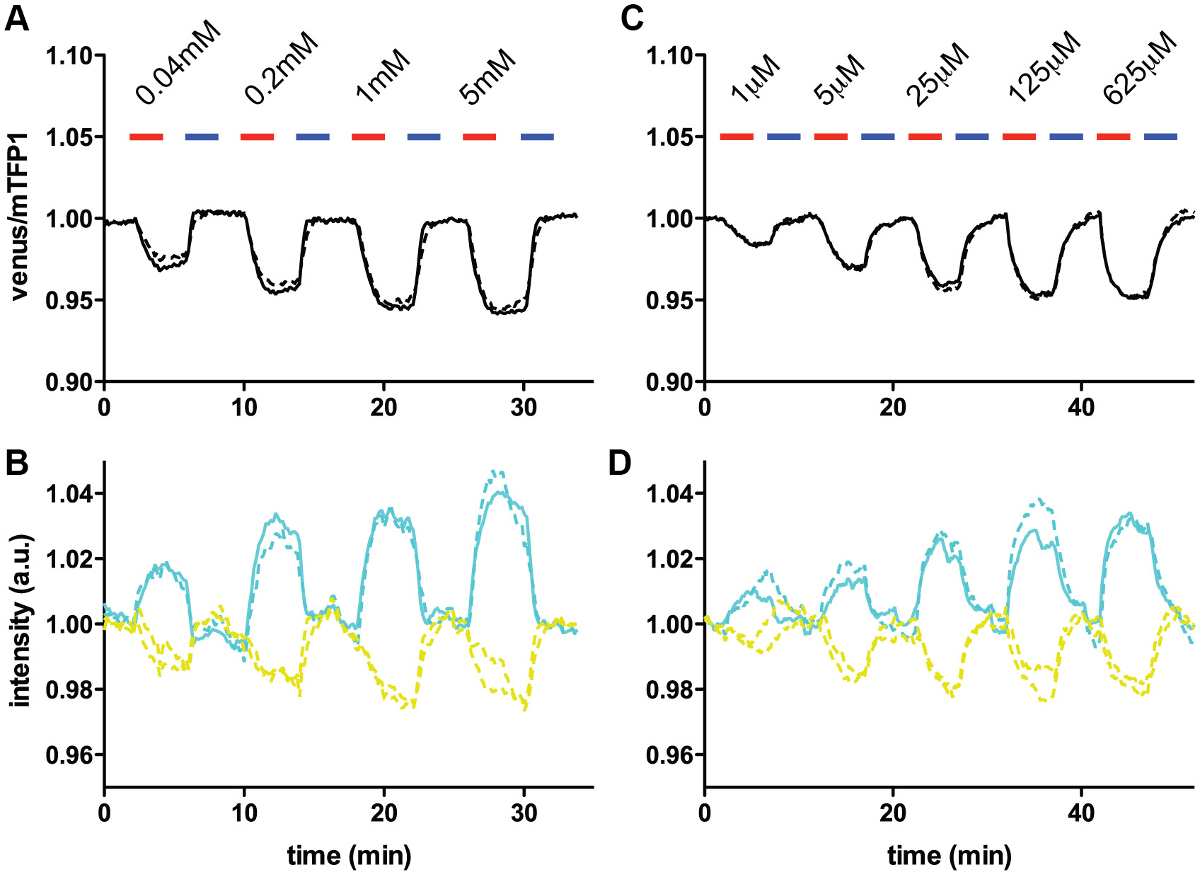
Figure 2. In vivo glutamine measurements using FLIPQ-TV3.0_8m and 100μ sensors. (A) The venus/mTFP1 ratio of cos7 cells co-expressing FLIPQ-TV3.0_8m sensor and ASCT2-mCherry. mCherry tag was used to identify the cells expressing the transporter without interfering the mTFP1 or venus emission channels. The cells were perfused with HEPES-buffered Hank’s buffer (pH 7.35). Timepoints when extracellular glutamine (red) and alanine (blue) was added to the perfusion media are indicated as boxes above the graph. Solid and dashed lines represent two individual cells measured in the same experiment. (B) The intensities of donor (mTFP1) and acceptor (venus) channels in the experiment shown in (A). The values were corrected for photobleaching and normalized to the baseline. (C) and (D) A similar experiment as in (A) and (B), performed with cos7 cells expressing the FLIPQ-TV3.0_100μ sensor. (Figure modified from 10).
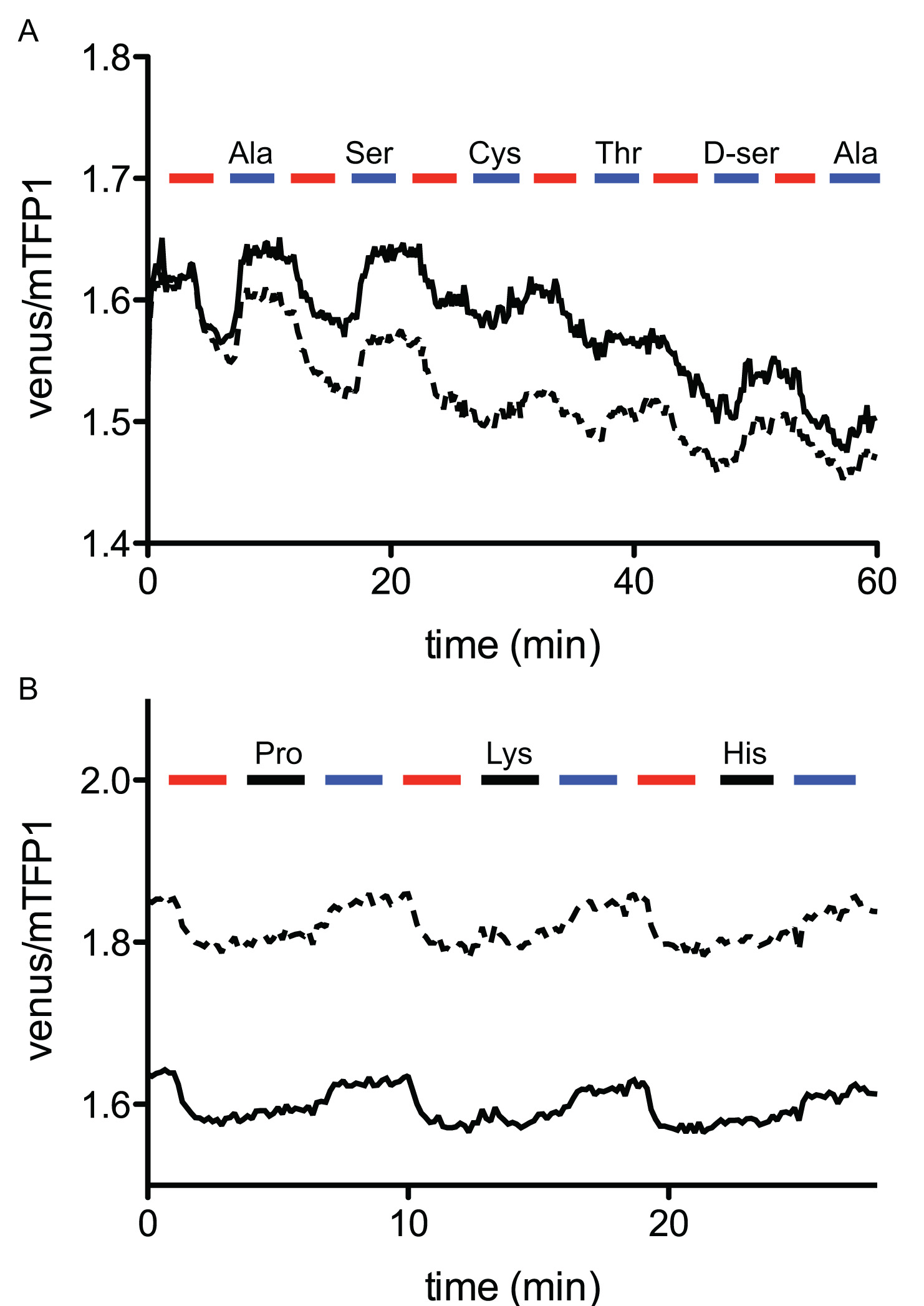
Figure 3. Elimination of cellular glutamine through the ASCT2 transporter in the presence of external amino acids, visualized using FLIPQ-TV3.0_8m sensor. (A) Cytosolic glutamine is exported by the addition of extracellular Ala, Ser, Cys, Thr, and D-ser. Timepoints when extracellular glutamine (red boxes) or other amino acids (blue boxes) were added to the perfusion media are indicated as boxes above the graph. (B) Addition of Pro, Lys, His (black boxes) does not alter cytosolic glutamine concentration, whereas the addition of Ala (blue boxes) promotes the export of glutamine. Solid and dashed lines represent two individual cells measured in the same experiment. All amino acids were added at 5 mM external concentrations. (Figure was originally published in 10).
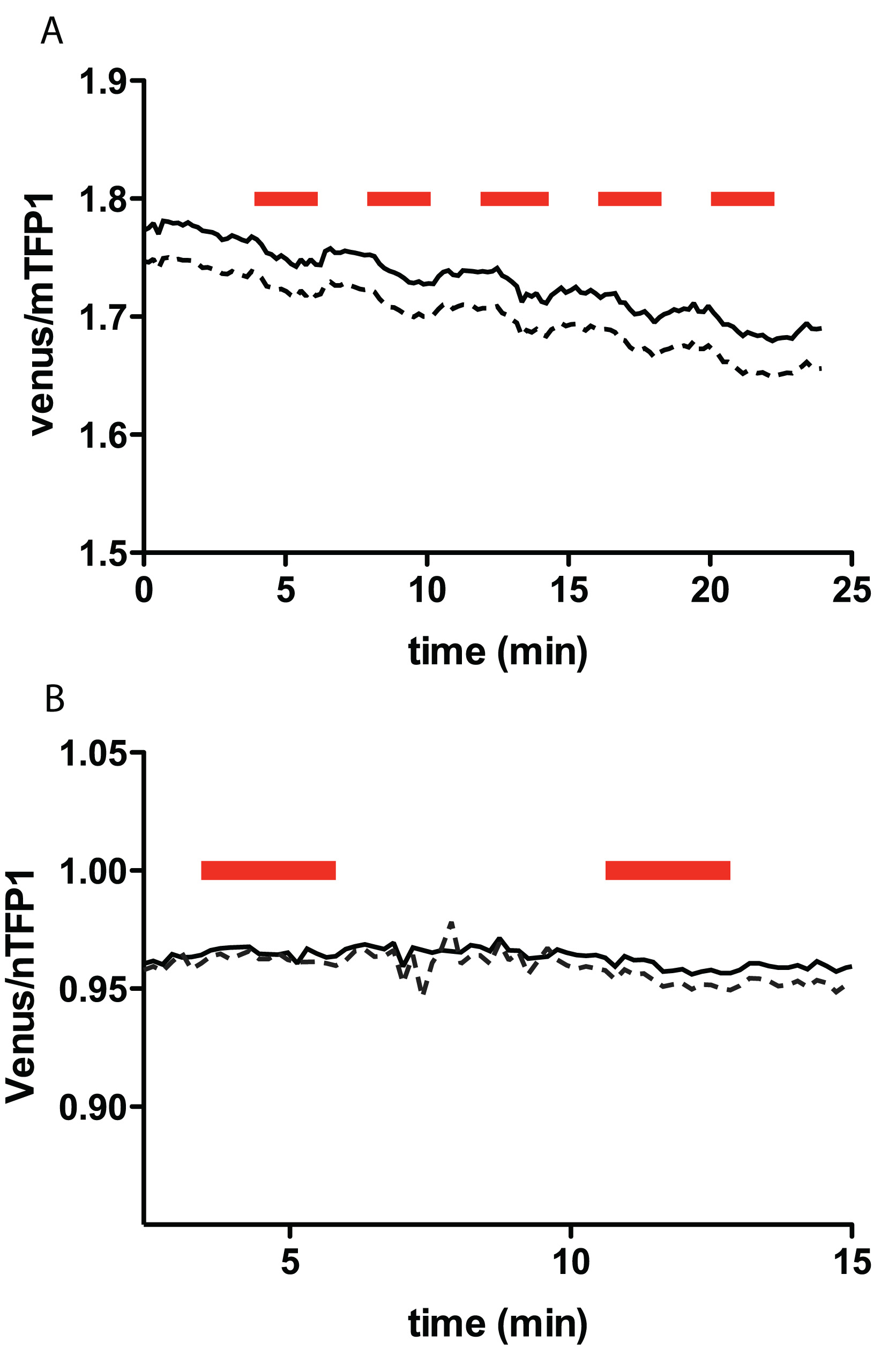
Figure 4. The venus/mTFP1 ratio of cos7 cells expressing FLIPQ-TV3.0_8m sensor (A) and 100μ sensor (B). The cells were perfused with HEPES-buffered Hank’s buffer. Timepoints when extracellular glutamine (5 mM) was added to the perfusion media are indicated as red boxes above the graph. Solid and dashed lines represent two individual cells measured in the same experiment.
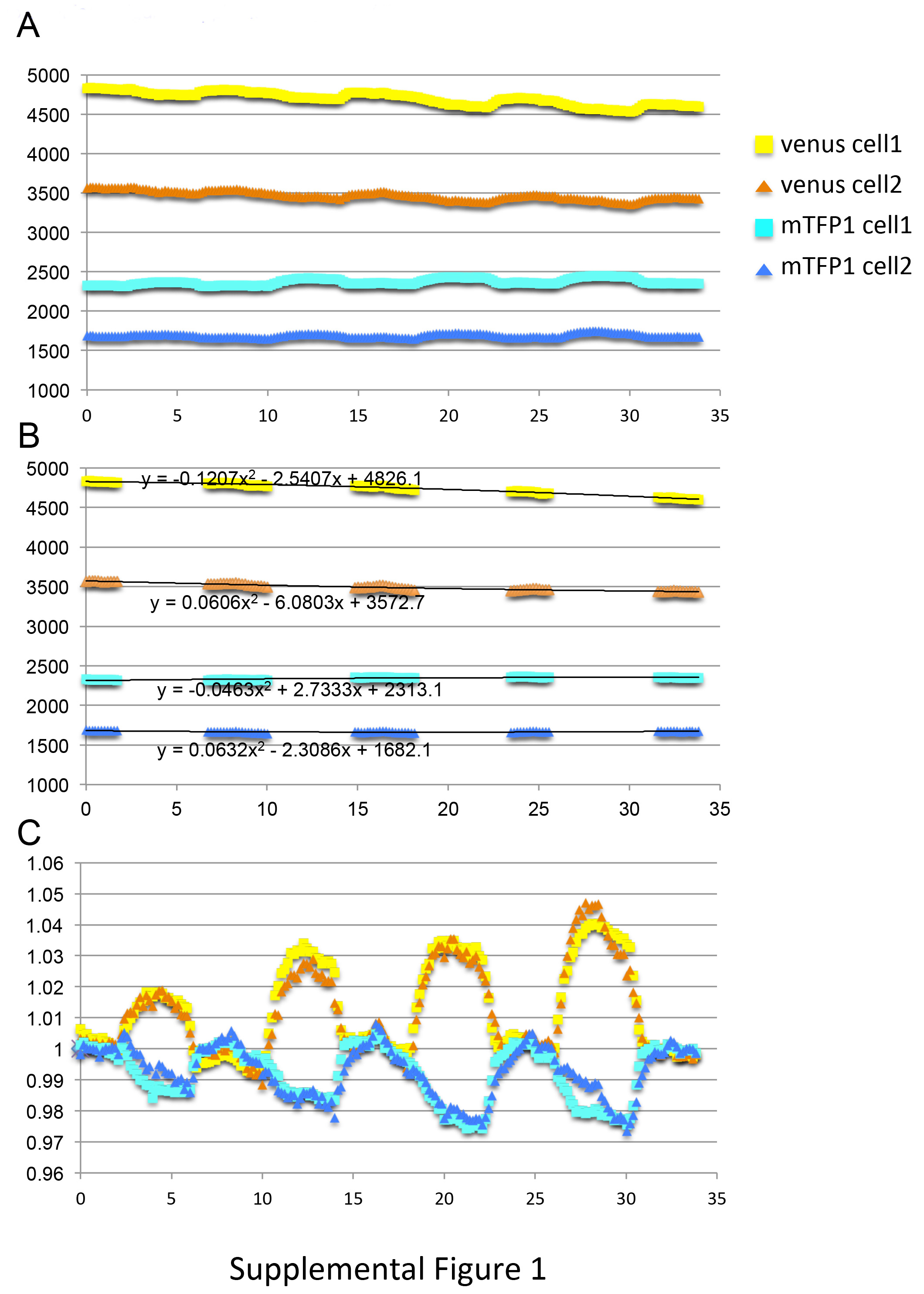
Figure 5. Correcting for photobleaching. (A) Raw intensities from two cells represented in Figure 2A and 2C. (B) Datapoints that were selected for calculating the baselines. Polynominal fitting curves are shown in the figure. (C) Intensities of channels shown in A, normalized against the baseline calculated in B. The dataset used in this figure is identical to the one shown in Figure 2A and 2C.
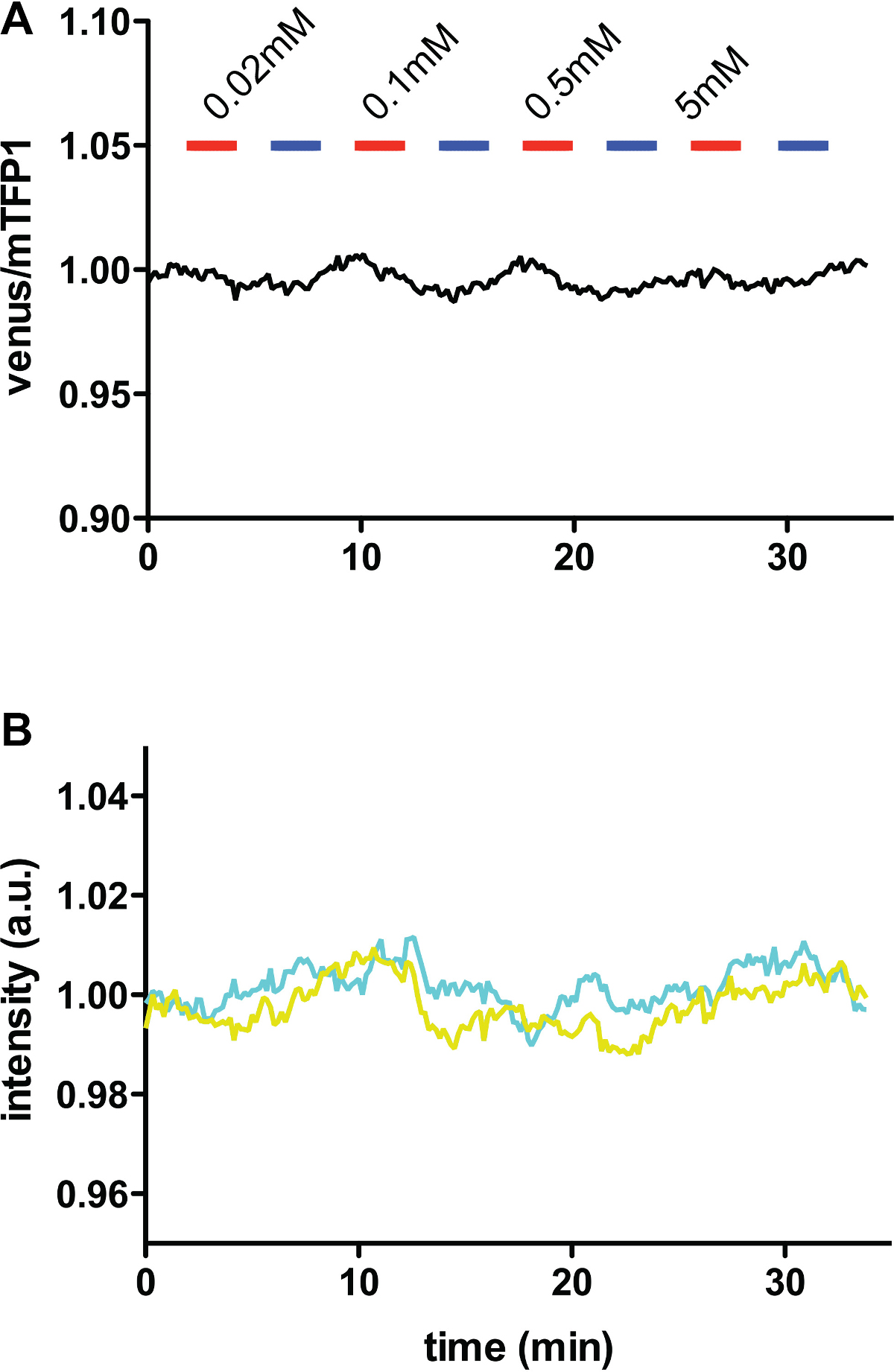
Figure 6. In vivo glutamine measurements using FLIPQ-TV3.0_1.5m sensor. (A) The venus/mTFP1 ratio of cos7 cells co-expressing FLIPQ-TV3.0_1.5m sensor and ASCT2-mCherry. mCherry tag was used to identify the cells expressing the transporter without interfering the mTFP1 or venus emission channels. The cells were perfused with HEPES-buffered Hank’s buffer (pH 7.35). Timepoints when extracellular glutamine (red) and alanine (blue) was added to the perfusion media are indicated as boxes above the graph. Solid and dashed lines represent two individual cells measured in the same experiment. (B) The intensities of donor (mTFP1) and acceptor (venus) channels in the experiment shown in (A). The values were corrected for photobleaching and normalized to the baseline.
| Time (min) | Solution A | Solution B | Solution C | Solution D | Solution E | Solution F |
| 0 | valve on | |||||
| 2 | valve off | valve on | ||||
| 4 | valve on | valve off | ||||
| 6 | valve off | valve on | ||||
| 8 | valve on | valve off | ||||
| 10 | valve off | valve on | ||||
| 12 | valve on | valve off | ||||
| 14 | valve off | valve on | ||||
| 16 | valve on | valve off | ||||
| 18 | valve off | valve on | ||||
| 20 | valve on | valve off | ||||
| 22 | valve off | valve on | ||||
| 24 | valve on | valve off | ||||
| 26 | valve off | valve on | ||||
| 28 | valve on | valve off | ||||
| 30 | valve off | valve on | ||||
| 32 | valve on | valve off | ||||
| 34 | valve off |
Table 1. Example of the perfusion protocol used in this experiment. Solution A: 9.7 g HANK salt (H1387), 0.35 g NaHCO3, 5.96 g HEPES to 1 L pH adjusted to 7.35 with NaOH. Solution B: Solution A + 0.04 mM Gln. Solution C: Solution A + 0.2 mM Gln. Solution D: Solution A + 1 mM Gln. Solution E: Solution A + 5 mM Gln. Solution F: Solution A + 5 mM Ala.
