NOTE: All tissue samples were obtained with approval of the institutional review board of San Raffaele Hospital (Milan, Italy).
1. Human Tissue Samples and Cell Purification (Figure 1)
NOTE: Leukemic lymphocytes were obtained from the Peripheral Blood (PB) of CLL patients, diagnosed according to Mulligan et al. 9
1.1) Leukemic Cell Separation from PB
- Collect PB in vacutainer blood collection tubes with Sodium Heparin.
- Transfer blood into a 15 ml tube and add human B-cell enrichment cocktail at 50 μL/mL of whole blood. Mix well and incubate 20 min at room temperature.
- Dilute sample with an equal volume of PBS + 2% FBS, mix gently and carefully overlay blood over the Ficoll, making sure not to break the interface. Use at least 1 part of Ficoll to 2 parts of the diluted sample (i.e. 4 ml Ficoll + 10 ml blood in a 15 ml tube).
- Centrifuge at 400 x g, at room temperature (20 °C) for 20 min with no brake. Mononuclear cells will be at the interface.
- Carefully aspirate the interface to recover the cells and place into a new tube. Wash cells once with PBS and centrifuge at 300 x g, 4 °C in the dark for 5 min. The pellet will be at the bottom.
- Discard the supernatant. Resuspend the pellet in the remaining supernatant by flicking the tube back and forth with fingers. Dilute the pellet with 10 ml complete RPMI medium (RPMI 1640 supplemented with 10% Fetal Calf Serum and 15 mg/ml gentamicin) and count cells using the Trypan Blue exclusion method.
1.2) Purity Evaluation
NOTE: Purity of all preparations needs to be always above 99%.
- Incubate 100 μl of purified cell suspension with the following antibodies (commercially available; see Table of Materials): CD3FITC (5 µl/test), CD14PE (5 µl/test), CD19ECD (6 µl/test), CD16-CD56PC5 (2 µl/test), CD5PC7 (4 µl/test), for 20 min at 4 °C in a FACS tube.
- Wash the sample with 4 ml of PBS (centrifuge for 5 min at 300 x g) and check the purity by flow cytometry.Check in the plot the % of CLL cells (> 99%) which co-express CD19 and CD5 on their surface as shown in Figure 1(6). Ensure that the preparations are virtually devoid of NK, T lymphocytes and monocytes by checking the % of CD3, CD14 and CD16-56 in the plot, which should be near zero.
1.3) Samples Storage
- For proteomic studies, collect 25 x 106 cells in a 1.5 ml tube, centrifuge cells at 300 x g for 10 min, discard the supernatant and lyophilize pellets for 4 hr. Keep at -80 °C until use.
- For validation assays, resuspend cells in complete RPMI (quantity depends on the further assay that is chosen) and proceed with Step 4.
2. Two-dimensional Electrophoresis (2-DE) (Figure 2)
2.1) Isoelectrofocusing (IEF)
- Solubilize CLL cells pellets with a final volume of 380 μl 2-DE buffer (344 μl of a RBthio solution (9 M Urea, 10 mM Tris, 4% CHAPS), 30 μl of 65mM DTT, 6 μl of 2% IPG buffer ampholine pH 4-7).
- Apply protein samples (1 mg) to 18 IPG strips (pH 4-7) and perform IEF following standard protocol as described by Conti et al. 10
- Equilibrate strips in 2 ml of Equilibration Buffer (EB: 50 mM pH 8.8 Tris-HCl buffer containing 6 M urea, 30% glycerol, 2% SDS), supplemented with 2% of fresh DTT, for 15 minutes at RT. Discard EB+DTT and add 2 ml of EB supplemented with 2.5% Iodoacetamide. Incubate for 15 min at RT on a rocking platform.
2.2) SDS-PAGE
- Dry the strips on a paper without touching the gel to remove the excess of EB. Load each strip on top of a 9-16% gradient SDS-PAGE gel. Add a small volume of SDS-Electrophoresis Buffer (Tris 25 mM, Glycine 192 mM, SDS 0,1% w/v) on top of the gel to facilitate loading of the strip.
- Seal the gel by adding ~5 ml of agar solution (0.8%) to prevent floating of the strip, then assemble the electrophoretic unit and fill with SDS-Electrophoresis buffer. Perform the run at 60 mA/gel for 4 hr at 4 °C.
- Remove the gel from the electrophoretic unit and place it in the proper staining box.
2.3) Silver Stain for Preparative Gel
| FIXER | 50% Methanol 12% Acetic Acid 0,05% Formalin |
2 hr (or overnight)* |
| WASH BUFFER | 35% Ehanol | 20 min (repeat the step three times) |
| SENSITIZING | 0,02% Sodium Thiosulfate | 2 min |
| WASH | H2O | 5 min (repeat the step three times) |
| SILVER NITRATE | 0,2% Silver Nitrate 0,076% Formalin | 20 min |
| WASH | H2O | 1 min (repeat the step twice) |
| DEVELOPER | 6% Sodium Carbonate 0,0004% Sodium Thiosulfate 0,05% formalin |
Until the staining is sufficient |
| STOP | 50% Methanol | 30 min |
| * 2 hr is the minimum time required for protein fixation |
Table 1.
NOTE: Protein spots can be visualized by staining gels with MS compatible silver stain11. All the solutions needed for silver staining are listed in Table 1.
- Prepare about 200 ml of each solution/gel and proceed as indicated in Table 1. Carefully add each solution to the staining box containing the gel. Perform all incubations on a rocking platform.
- After the staining, remove the Stop Solution and leave the gel in a solution of 1% Acetic Acid until acquisition.
2.4) Gel Acquisition and Analysis
- Acquire images at high resolution using a densitometer within 3 days from the staining.
- Analyze 2-D protein patterns by using software for 2-DE gels.
NOTE: The software allows a fast and reliable image comparison.- Create a project by choosing “create a new project” from the contextual menu. Create a folder and import the gels by choosing “import gel images” with .tif, .png. or .gel extension.
- Once the gels are imported and added to the workspace, perform an automated spot detection by selecting the gels for spot detection and choosing “edit > spot> detect” in the menu.
- Next, perform a background subtraction by selecting from the menu “subtract background”. Note: After that it is possible to compare multiple gels and to detect differences or similarities by quantitative and/or qualitative analysis.
3. Protein Identification by MALDI-TOF MS analysis (Figure 3)
3.1) Protein Digestion
- Rinse the gel with water and excise spots of interest from gels either by manual (cutting with a clean scalpel) or by automated excision.
- Wash gel particles with water (100-150 μl), spin down (30 sec at 400 x g) and remove the liquid.
- Add an equal volume (depending on the gel volume) of CH3CN to the gel pieces and wait for 10-15 min until the gel pieces shrink. Dry the gel particles in a vacuum centrifuge.
- Add 75-100 µl of 10 mM Dithioerythritol diluted in 50 mM NH4HCO3 and incubate for 30 min at 56 °C (Reduction Step). Shrink again the gel pieces with CH3CN as described in Step 3.1.3.
- Proceed with alkylation by adding 75-100 µl of a solution 55 mM Iodoacetamide diluted in 50 mM NH4HCO3 and incubate for 20 min at room temperature in the dark.
- Shrink the gel pieces once more with CH3CN using enough volume to cover the gel pieces as described in Step 3.1.3. Dry in a vacuum centrifuge.
- Rehydrate the particles in a buffer containing 25 mM NH4HCO3, 5 mM CaCl2,and 12,5 ng/μl of trypsin at 4 °C for 20 min. Use enough volume of buffer to cover the gel pieces. Remove the un-absorbed supernatant from the gel pieces and add 25 mM NH4HCO3, 5 mM CaCl2 without trypsin to cover the gel.
- Leave samples at 37 °C at least 3 hr (the minimum time required for peptide digestion) to overnight.
3.2) Mass Spectrometry Analysis (Dried droplet technique)
- Spot 1 μl aliquot of each sample onto a stainless steel MALDI plate and mix with 1 µl of α-cyano-4-hydroxycinnamic acid as matrix, prepared in 50% CH3CN and 0.1% TFA.
- Obtain mass spectra on a MALDI–TOF mass spectrometer. Accumulate about 200 spectra per spot, adjusting laser intensity to prevent signal saturation or to increase the signal. Process spectra via Data Explorer software as described below:
- Import the .dat file and perform the baseline correction by choosing “process > baseline correction”. Calibrate masses using the trypsin autolysis products and matrix peaks by choosing “process > mass calibration > manual calibration; select peaks close to m/z 855.1 and 2163.1 by left dragging and select the corresponding reference masses, then click “plot” and “apply calibration” in the manual calibration window.
- Adjust the threshold for peak detection (“peak >peak detection”), duplicate the active trace (“display >duplicate active trace”), select the new trace and perform the deisotoping (“peak >deisotoping”). Using the macro “getpeaklist”, generate a list of masses to be used for the identification. If required, manually remove noise signals picked as peaks to clean the list and get a better identification.
- Identify proteins by searching a comprehensive non-redundant protein database using ProFound and Mascot as search engines12.
- Use the following parameters in all searches: one missed cleavage per peptide, methionine oxidation as variable modification, cysteine carbamidomethylation as fixed modification and an initial mass tolerance of 50 ppm.
4. Cytoskeletal Activity Assays (Figure 4)
NOTE: Resuspend cells purified in Step 1 in complete RPMI (106 cells/200 μl).
4.1) Migration Assay. This test allows quantification of the migratory capacity of the analyzed cells (lymphocytes ). Use a transwell chamber of 6.5 mm diameter and 5.0 μm pore size and perform the assay in triplicate. Optimize pore size and migration time (Step 4.1.3) in the case of different cell types.
- Prepare the lower chamber by adding 600 μl of complete RPMI with or without the specific stimuli (i.e. SDF-1) to measure the induced and the spontaneous migration respectively.
- Put the upper chamber on it and let the system equilibrate for 30 minutes at 37 °C in the incubator.
- Seed 106 cells/200 μl in the upper chamber (total cell number) and incubate the plate for 4 hr at 37 °C in the incubator.
- Remove the upper chamber, collect the media in the lower chamber and wash the lower chamber once with 1 ml PBS. Collect the PBS in the tube.
- Centrifuge 5 minutes at 300 x g, discard the supernatant and resuspend in 500 μl PBS.
- By flow cytometry, count the number of migrated cells after 1 min of acquisition. In case of an isolated cell population it is not necessary to add any surface antibody. Check the number of cells in a bi-dimensional dot plot considering the side scatters of the cells and the number of events visualized in 1 min, which is the number to use for calculation.
- Calculate the Migration Index (MI) as:

4.2) Adhesion assay. This assay allows measuring the adhesion capacity of the cells. Perform the assay in triplicate.
- Pre-coat 96-wells plate (flat-bottom) with 50 μl/well of either 2% BSA-PBS (as background) or 1 μg ICAM-1 diluted in PBS, overnight at 4 °C.
- Wash the plate twice with PBS and block non-specific sites by adding 2% BSA-PBS (100 μl/well) for 1 hr at 37 °C.
- In the meantime resuspend cells at 5 x 106/ml in complete RPMI, label them with 1 mM CMFDA-green cell tracker and incubate 30 min at 37 °C.
- Add 1 x 106 cells/well to the pre-coated wells after discarding the blocking solution (Step 4.2.2) and incubate 1 hr at 37 °C.
- Quantify adhesion by using an ELISA reader (Excitation filter: 485 nM, Emission filter: 535 nM). Perform a first measurement on the total input (INPUT).
- Wash the plate gently with 2% BSA-PBS (200 μl/well) 4 times (x). Perform a reading of the plate after each washing step (Adhesionx).
- After background subtraction for each measurement, calculate specific adhesion (ADHESION):
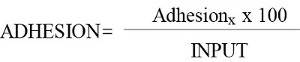
4.3) Polymerization assay. This is a colorimetric assay that quantifies F-actin polymerization. Perform the assay in triplicate.
- Resuspend 1×10^6 cells in 100 μl of pre-warmed complete RPMI and add the specific stimulus (i.e. α-IgM 20 μg/ml ) for 10 min.
- Stop the reaction with 400 μl of 4% paraformaldehyde and fix cells for 10 min at RT.
- Wash 3 times with PBS (centrifuge at 300 x g for 5 min, 4 °C).
- Discard the supernatant and permeabilize cells with PBS-saponine 0.2% (200 μl) on ice for 2 min.
- Wash 3 times with PBS-saponine 0.1% (centrifuge at 100 x g for 5 min, 4 °C), discard the supernatant and resuspend in 100 μl PBS-saponine 0.1%.
- Stain with Phalloidin-Alexafluor 488 (3 μg/ml) for 30 min at room temperature in the dark.
- Wash 3 times with PBS-0.1% saponine. Discard the supernatant and resuspend in 300 μl PBS.
- Quantify the amount of F-actin by flow cytometry, calculating the Mean Florescence Intensity (MFI) of the Phalloidin. Analyze the generated data in a single dimension plot in order to produce a histogram based on the fluorescent intensity, the MFI value is shown in the graph report.
- Measure F-actin polymerization (∆ F-actin) as:
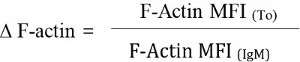
We isolated primary leukemic B cells form PB of CLL patients and we analyzed proteomic maps. Samples (n = 104) were grouped in two main subsets based on the clinical features of each patient (bad prognosis vs good prognosis) and comparative analysis of 2DE gels was performed.
The analysis allowed identification of spots differentially expressed between the two subsets (in term of presence/absence or shift on the gel, implying changes in post-translational modifications; n ≈ 16). We excised selected spots from the 2DE gels and after being reduced and alkylated, proteins were digested with trypsin and peptides in the resulting supernatant were spotted onto a MALDI plate. Spectra were acquired in a MALDi-ToF Voyager DE. The resulting peak list was submitted to MASCOT and ProFound. Masses within a certain mass tolerance are assigned to peptide sequences in the database, and then assembled into a protein, which is considered identified if it passes a certain probability score (Figure 3). By this analysis we identified proteins mainly involved in cytoskeletal activity and metabolic processes (unpublished data).
Among them, we focused on hematopoietic-lineage-cell-specific-protein-1 (HS1) whose differential phosphorylation strongly associated with the clinical course of the disease (Figure 4). In particular, we have found that patients carrying a single spot (n = 44, hyper-phosphorylated HS1) experience a bad clinical outcome, while patients with 2 spots (n = 60, hypo-phosphorylated HS1) have good prognosis13 (Figure 4). The presence of the HS1 protein in the spots was then validated by immunoblotting the 2DE gel with a monoclonal antibody against HS1 13.
Since it is known that HS1 is involved in cytoskeletal remodeling14, we performed in vitro assays to test if the HS1 phosphorylation status could differentially affect cytoskeletal activity in the two CLL subsets (good vs bad prognosis). We found that CLL cells carrying HS1 as one spot have an impaired cytoskeletal activity in terms of migration, adhesion and actin polymerization, compared to hypo-phosphorylated-HS1 samples, thus explaining a different clinical behavior15 (Figure 5).
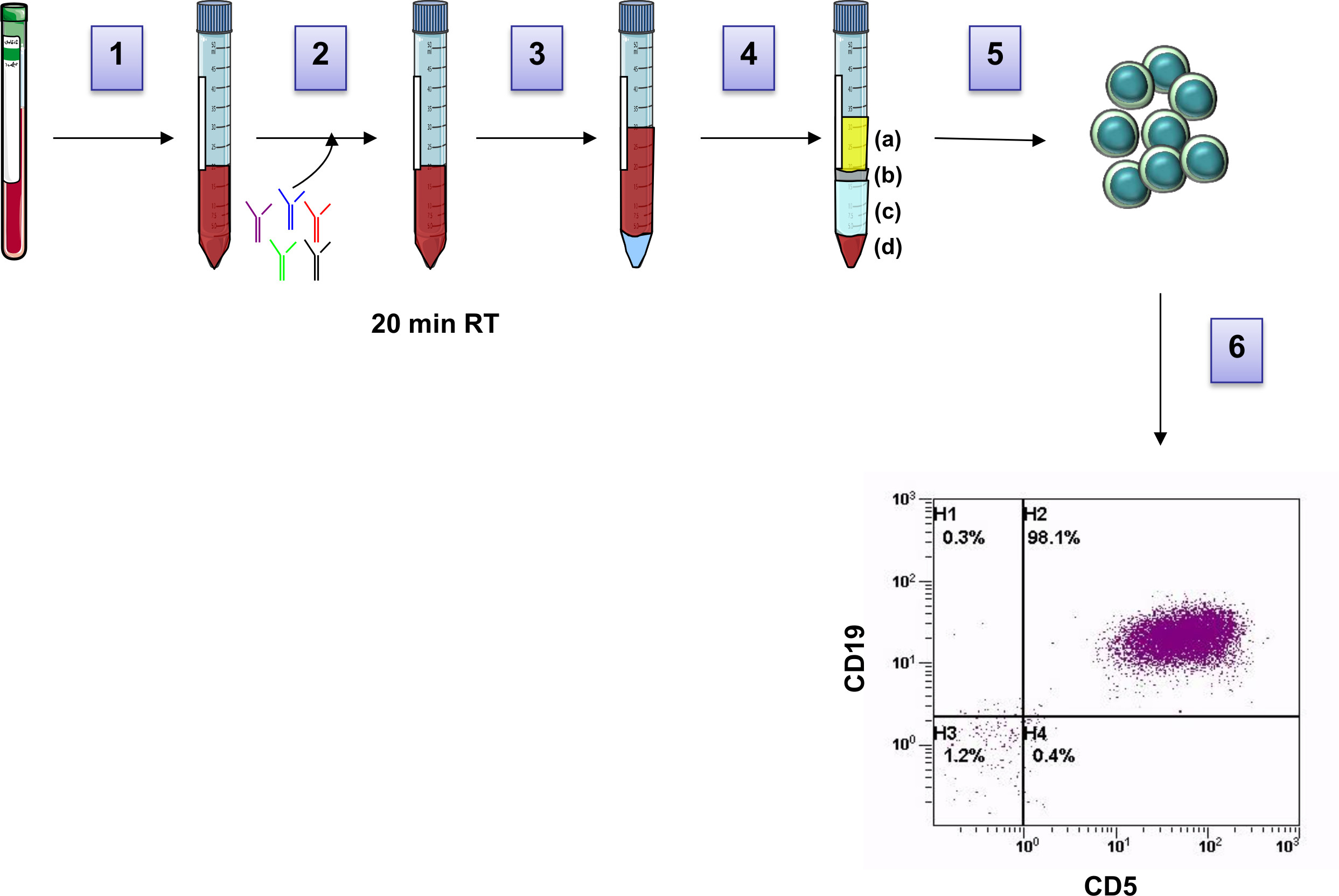
Figure 1: Workflow of leukemic cell purification from PB. Blood from CLL patients is transferred into a 15mL tube (1), Human B-cell enrichment cocktail is added to the sample (2) and incubated for 20 minutes. Blood is then diluted with PBS in a proportion of 1:1 and laid on the top of the density gradient (3). Subsequent sample centrifugation (4) allows the formation of multiple layers: a) plasma, b) B lymphocytes, c) Ficoll, d) red cells and unwanted cells. Purified B lymphocytes (b layer) are then collected (5), washed and purity of cell preparation is analyzed by flow cytometry (6). Please click here to view a larger version of this figure.
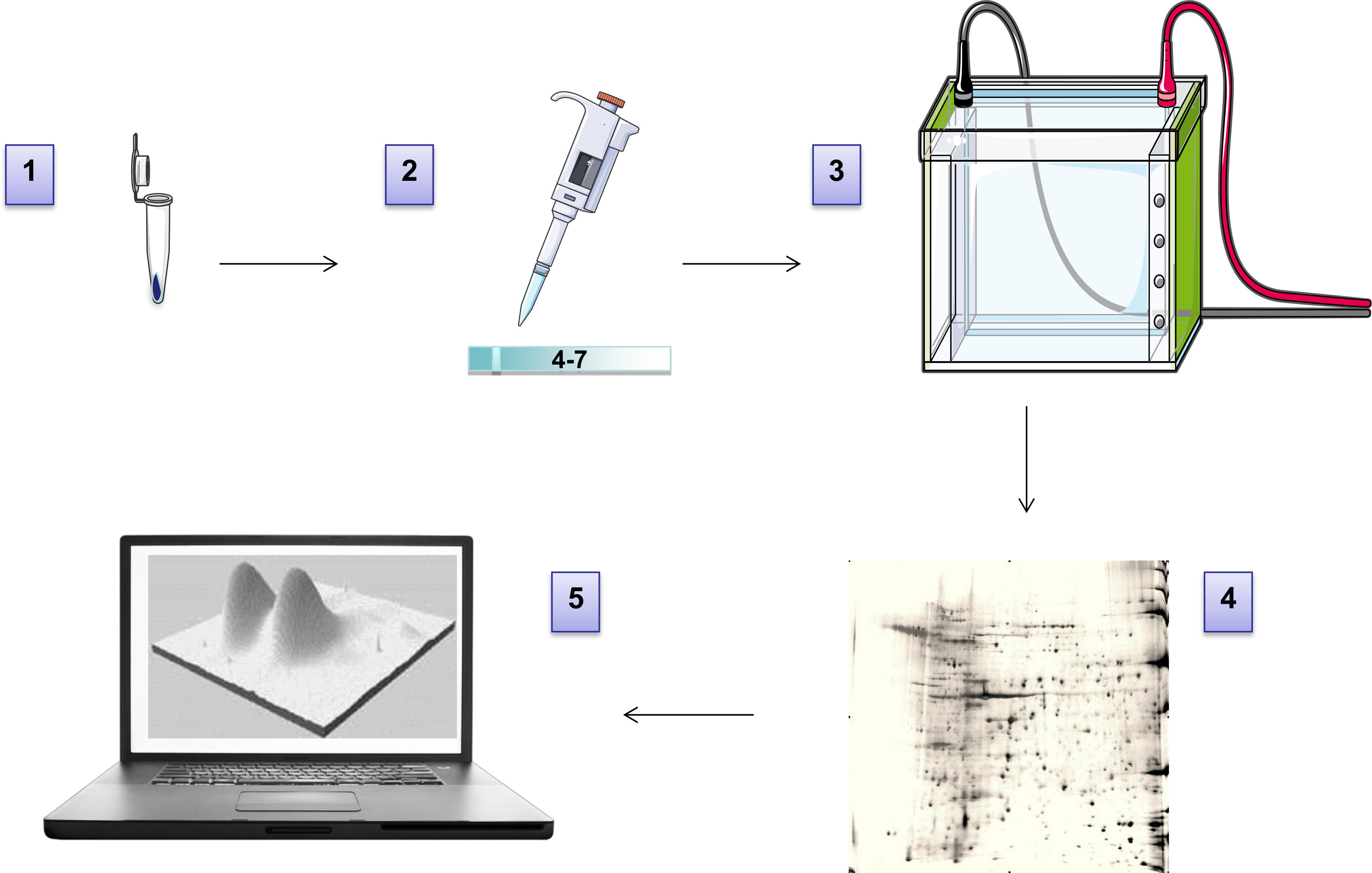
Figure 2: Workflow of 2DE. The pellet is solubilized in 2DE buffer (1) and loaded on the IPG strip for the first dimension run (2-isoelectrofocusing). After equilibration, the strip is loaded on top of a gradient polyacrylamide gel for the second dimension run (3-SDS-PAGE). The gel is then stained to visualize spots/proteins (4). After high-resolution image acquisition, proteomic maps are analyzed (5). Please click here to view a larger version of this figure.

Figure 3: Workflow of MS analysis. (1) Spots of interest are excised from the 2D-gel with a scalpel and transferred into a clean tube. (2) After being reduced and alkylated, proteins are digested with trypsin, peptides in the resulting supernatant are spotted onto a MALDI plate (3). Spectra are acquired in a MALDi-ToF Voyager DE. (4) The resulting peak list is submitted to MASCOT and ProFound search engines and searched against a comprehensive non-redundant protein database. (5) Masses within a certain mass tolerance are assigned to peptide sequences in the database, then assembled into a protein. Please click here to view a larger version of this figure.

Figure 4: HS1 phosphorylation status correlates with prognosis of CLL patients. (1) The circle identifies two close spots with the same molecular mass (Mr) of 79 kDa and different isoelectric point (pI) of 4.83 and 4.86 respectively, which was identified by MS as HS1 protein (2). (2) Two representative gels of one bad prognosis (red square) and one good prognosis CLL patients (green square). (3) Kaplan-Meier curves show cumulative survival of CLL patients grouped according to HS1 phosphorylation pattern (1 spot, n = 44, vs 2 spots, n = 60). Patients with 2 spot (green dots) have a significantly longer survival (median survival not reached) than those with only one (red dots). Please click here to view a larger version of this figure.

Figure 5: HS1 phosphorylation status influences cytoskeletal functionality in CLL primary samples. (1) Migration on transwell of primary samples in the presence or absence of SDF-1. In the graph are displayed the means SEM of the number of cells acquired in 1 minute at the flow cytometer (n = 7 of 1 spot HS1 vs n = 12 of 2 spot HS1). (2) Spontaneous adhesion was measured after cell labeling and 1 hr incubation in 96-well plates. Displayed are the means SEM for primary samples (n = 7 of 1 spot HS1 vs n = 12 of 2 spot HS1). (3) F-actin polymerization capability of primary samples. Displayed are the means SEM of the relative F-actin content of CLL cells after stimulation with SDF-1 and staining with FITC-labeled phalloidin (n = 5 of 1 spot HS1 vs n = 5 of 2 spot HS1). MFI: Mean Fluorescence Intensity. The histogram represent as example of sample acquisition. Please click here to view a larger version of this figure.
