As an initial proof of concept, purified green fluorescent protein was successfully introduced into mammalian cells using electroporation. GFP, an approximate 27 kD a molecular weight protein is commonly introduced into mammalian cells (normally expressed from plasmid DNA) as a molecular biology tool without significant cellular toxicity. HeLa cells were incubated (Figure 1A) or electroporated (Figure 1B) with 25 µg/ml GFP, followed by immunofluorescence confocal microscopy to check for fluorescent GFP signal. To demonstrate that the GFP was inside the cellular cytosol, the cells were also stained with wheat germ agglutinin (WGA) to delineate the cytosolic boundary (red) as well as a nucleic acid stain, DAPI (blue) to indicate the nucleus. Intracellular GFP signal was observed only upon electroporation, while no intracellular protein was observed in cells incubated with GFP. Similar results were observed with RAW264.7 mouse macrophage-like cells (Figure 1C and 1D). Note that WGA is a lectin that preferentially binds to residues in the plasma membrane and thus can exhibit punctate staining based on the length of incubation. Compare Figure 1A and Figure 2A, where Figure 2A was incubated for a longer duration than Figure 1A.
Electroporation was then extended to a tagged, purified Salmonella effector, GtgE. GtgE, a known virulence factor 30, was discovered by our group to be secreted into host cells 31, and was recently shown to be a cysteine protease 32. HeLa cells were incubated or electroporated with 50 µg/ml GtgE. For immunofluorescence analysis, the cells were stained with an antibody against the streptavidin-binding peptide affinity tag on GtgE. The cells were also stained with WGA and DAPI. In incubated HeLa cells (Figure 2A), there is no intracellular GtgE as visualized by a lack of green fluorescent foci. In contrast, electroporated HeLa cells (Figure 2B) show significant fluorescent intracellular signal indicating effector protein had entered the cells due to the electroporation process. RAW cells showed a slightly increased propensity to accumulate protein on the cellular surface during incubation (Figure 2C), but intracellular signal was seen only upon electroporation (Figure 2D).
Determining the limits of detection for visualizing sub-cellular localization via confocal microscopy was important to ensure enough protein entered the cell, while avoiding overloading the cell with potentially toxic bacterial effectors. In Figure 3, GtgE was electroporated into RAW macrophage-like cells at 2.5, 25, and 50 μg/ml. The cells were then fixed and stained with an antibody against the tag on GtgE. Only a very small number of foci were observed at 2.5 μg/ml GtgE (Figure 3A), with increased numbers of foci apparent at 25 μg/ml, (Figure 3B), but the greatest number of distinct foci were seen at the maximum protein concentration tested, 50 μg/ml (Figure 3C). Similar staining patterns were observed between the samples indicating at these protein concentrations intracellular protein could be easily verified by confocal microscopy. Protein concentrations above 50 μg/ml were not tested because as the concentration of protein increased, so did the propensity for the target protein to become adsorbed to the surface of the cells. To avoid these aggregates of membrane associated protein, it became necessary to pool two electroporation cuvettes to obtain enough host interacting protein to visualize on a western blot (Figure 6, far right lane).
To further demonstrate that the introduced protein was not simply adsorbed to the surface of the cell, consecutive optical sections were imaged at focal planes every 0.35 to 0.43 micrometers (Z-stacks) using confocal microscopy. RAW cells were electroporated with 50 µg/ml GtgE and stained as described above (Figure 4, A and B). The Z-stacks showed that GtgE was indeed intracellular and the foci extend from the bottom (Figure 4A, plane 6 of 36) to the top of the cell (Figure 4B, plane 26 of 36). Similar results were obtained for all electroporated proteins. The intrinsic fluorescent profile of control cell populations with and without effector protein or electroporation was examined by fluorescent confocal microscopy, and was found to be negligible.
Additionally, the electroporated protein was examined to see if it was targeted for degradation via endocytic pathways. Cells were stained for Ras-related protein 5A (Rab5), a marker for early endosomes (Figure 4C), or Lysosome-associated membrane protein 1 (Lamp1), a marker for late endosomes and lysosomes (Figure 4D). Co-localization between the electroporated protein and these cellular markers would indicate a close physical interaction between them (i.e. that the electroporated protein was inside the endosomes/lysosomes). Confocal microscopy showed that the electroporated protein (GFP or GtgE) did not co-localize with LAMP1 or Rab5. It was inferred from this that introduced protein was not directly endocytosed into the cells or targeted to the endocytotic pathway within four hours of treatment.
Exogenous protein introduction can have cellular effects over the course of several hours or days, and thus it is often beneficial to examine the persistence of introduced proteins. To determine how long an electroporated protein would persist without degradation after electroporation. Based on visualization of attachment and cell spreading, 4 hr was determined to be the approximate minimum recovery time for electroporated cells. To temporally observe protein persistence a time-course experiment was performed, staining the cells 4, 24, or 96 hr after electroporation. After 4 hr (Figure 5A), when cell morphology suggested recovery, there was an appreciable amount of intracellular protein. After 24 hr (Figure 5B), there was still substantial electroporated protein inside the cells. Even when the time after electroporation was extended to 96 hr before fixation and staining (Figure 5C) there were observable foci albeit at a reduced apparent abundance, demonstrating protein persistence days after initial treatment.
Two important caveats were noted during the course of these experiments. RAW cells exhibited a greater tendency than HeLa cells to accumulate exogenous proteins on the cellular surface irrespective of incubation (Figure 6A) or electroporation (Figure 6B). Despite this, all electroporated samples had increased internal protein (Figures 1, 2, 5B). Additionally, the membrane-associated protein disappeared over time. A second caveat was a small population of cells exhibiting a phenotype consisting of smaller, rounded cells loaded with high amounts of exogenous proteins in both control (incubated) and treated (electroporated) cells (Figure 6 C and D, incubation samples shown). The nuclei of these cells showed condensed chromatin based on DAPI staining, and filled the entirety of the cellular volume, suggestive of apoptosis. It is therefore possible that apoptotic cells (arising as a normal part of the cell cycle or due to harvest/treatment) have a propensity to absorb protein from the buffer.
To demonstrate that electroporated proteins were functional and could localize correctly inside the host cell, co-localization and protein-protein interaction between the Salmonella effector protein SspH1 and its known host target, protein kinase N1 (PKN1) 33 was shown. After electroporation, the physical interaction between SspH1 and PKN1 was verified by an affinity based immunoprecipitation followed by western blot (Figure 7A). Co-localization was also observed by confocal microscopy, which indicates the fluorophores are close enough spatially to overlap 34. After electroporation with 50 µg/ml SspH1, HeLa cells (Figure 7B) were fixed and stained for SspH1 (green) and PKN1 (red). At low laser power distinct foci (yellow) were observed in the nucleus indicating SspH1 and PKN1 were physically close enough to interact. This interaction has been established in the literature; however this study is the first to show co-localization in the nucleus.
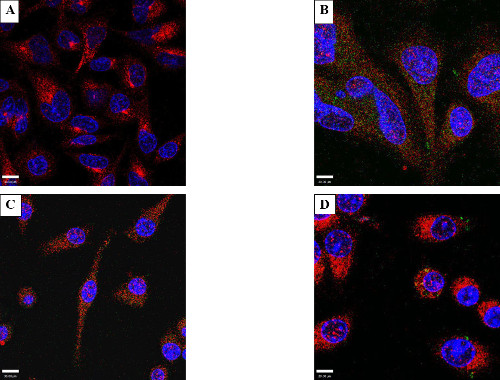
Figure 1: Electroporation of GFP – HeLa or RAW 264.7 cells. Cells were incubated or electroporated with 25 µg/ml of purified green fluorescent protein (GFP) and stained with anti-GFP antibody (Green), DAPI, a nuclear specific probe (Blue), and WGA (Red) to delineate the cytosolic boundary. Incubated (A) HeLa cells show an absence of green fluorescence indicating a lack of internalization of GFP. (B) shows a representative photomicrograph of electroporated GFP. Note the appearance of intracellular foci indicating GFP had entered the cells. (C) and (D) are representative images of RAW cells incubated (C), or electroporated (D) with 25 µg/ml of purified green fluorescent protein.
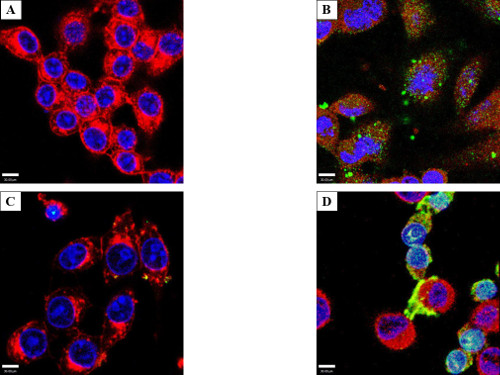
Figure 2: Electroporation of Salmonella effector GtgE – HeLa cells. Cells were incubated (A) or electroporated (B) with 50 µg/ml of an affinity tagged Salmonella effector, GtgE, and stained with an anti-effector tag antibody (Green), DAPI, a nuclear mask (Blue), and WGA (Red) to delineate the cytosolic boundary. In (A), incubated HeLa cells show an absence of green fluorescence indicating a lack of internalization of GtgE. (B) shows a representative photomicrograph of electroporated GtgE, showing intracellular electroporated effector protein. (C) and (D) are representative images of RAW cells that were either incubated or electroporated, respectively, in the same fashion with 50 µg/ml of Salmonella GtgE. Light blue is the combination, or overlay, of red fluorescence from the WGA and green fluorescence from the secondary antibody.
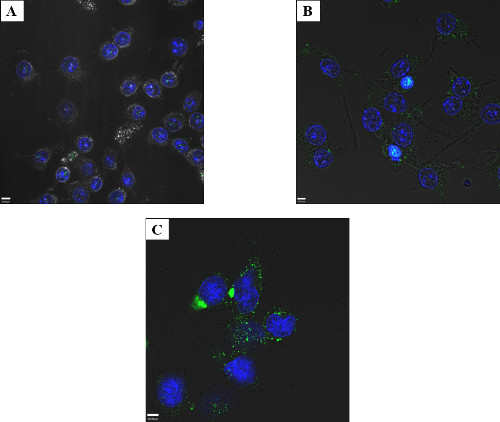
Figure 3: Titration of electroporated protein – RAW macrophage-like cells. Cells were electroporated with 2.5 μg/ml (A), 25 μg/ml (B), or 50 μg/ml (C) Salmonella effector GtgE and allowed to recover for 4 hr. The cells were fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). It is possible to visualize fluorescent foci, indicative of intracellular GtgE at 2.5 μg/ml via confocal microcopy, although better results were obtained when higher starting amount of protein were used. Satisfactory results (including intracellular protein easily observed by confocal microscopy without excessive membrane associated aggregates) were obtained at 50 μg/ml for GtgE and thus no greater concentrations were tested.
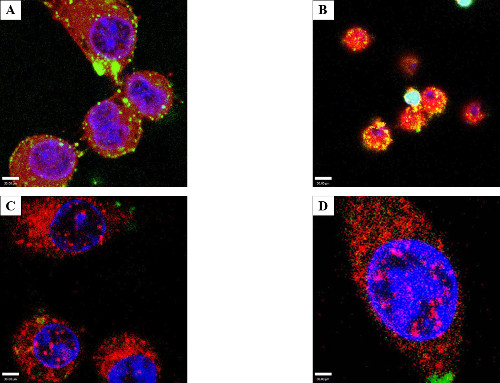
Figure 4: Protein internalization and lack of co-localization with endosome markers. Consecutive optical slices (z-stacks) obtained by confocal microscopy to visualize if the electroporated protein was internal. RAW cells were electroporated with 50 µg/ml GtgE. (A) shows optical slice 6 of 36, the z-stack (2.1 micrometers) above the bottom of the dish. (B) shows the same field of view but at slice 26 of 36. The intracellular foci extend throughout the cell indicating that the protein is truly intracellular and not aggregated on the surface of the cell. Light blue is the combination of red WGA, green secondary antibody, and blue DAPI. Co-localization assay with markers of endosomes/lysosomes, Rab5, (C) or a lysosome marker, LAMP1 (D). The effector protein has been stained green, the markers (Rab5 or LAMP1) stained red, and the nuclei delineated with DAPI, blue. There is no evidence of co-localization with either LAMP1 or Rab5 indicating that the protein was not phagocytized.
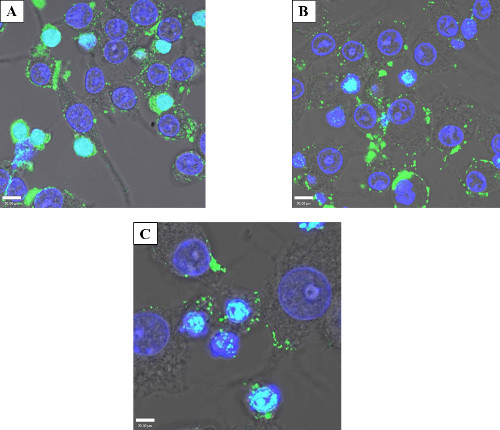
Figure 5: Protein persistence after electroporation. Confocal micrograph showing the persistence of electroporated GtgE over time. 50 µg/ml GtgE was electroporated into HeLa cells. The cells were then fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). 4 hr (A) was determined to be the minimum amount of time to allow cells to recover and as expected shows maximum intracellular protein. After 24 hr (B) and 96 hr (C), green foci, both intracellular and on the cellular surface, show effector persistence indicating that the protein is not degraded days after the initial treatment. The light blue color in this image is the overlay of blue DAPI and the green antibody staining.

Figure 6: Cell surface protein aggregation and an electroporation phenotype. RAW macrophage-like cells were either incubated (A) or electroporated (B) with 50 µg/ml GtgE and stained with anti-SBP-tag antibody (Green), WGA (Red), and with DAPI (Blue). Both RAW 264.7 and to a lesser degree HeLa cells, tended to aggregate effector on the surface of the cell; all electroporated cells were shown to have increased internal protein (HeLa not shown). Yellow is the overlay of red from WGA and green from target protein. RAW (C) and HeLa (D) cells showing an altered phenotype after incubation with GtgE (shown). Several experiments showed a phenotype consisting of smaller cells with differential DAPI staining and a loss of cytoplasm. Light blue is the overlay from the red WGA, green secondary antibody, and blue DAPI. Confocal microscopy was used to show the target protein accumulating in the nucleus. Since this phenotype of exogenous protein-laden cells appeared after both incubation and electroporation, one could hypothesize this phenotype to be suggestive of apoptosis.
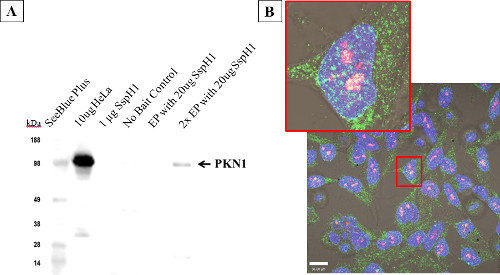
Figure 7: Host protein interactions with electroporated Salmonella SspH1 – HeLa cells. Cells were electroporated at the listed concentrations with SspH1, allowed to recover for 4 hr before performing a modified affinity purification followed by western blot (A) to enrich for and detect the known eukaryotic interacting partner: serine/threonine protein kinase N1 (PKN1). Note that the far right lane (2x EP) includes 2 pooled cuvettes, as the signal from one cuvette blended into background, yet PKN1 was still enriched above the lack of binding seen in the no-bait control. The western blot was run with native HeLa lysate, purified SspH1, and the affinity purification samples before being probed with a monoclonal antibody to PKN1. Target detection was obtained via a horseradish peroxidase conjugated secondary antibody with reactivity against the isotype of the primary antibody. HeLa cells (B) showing co-localization (yellow) between PKN1 (red) and SspH1 (green) via by confocal microscopy after electroporation with 50 µg/ml SspH1. This optical overlap indicates that the effector and the host protein are close enough to physically interact. The fact that the interaction was shown in the nucleus for the first time, offers additional support that the protein was trafficked correctly by the host.
