A representative result of total RNA and amplified aRNA from Day 7 bovine embryos is shown in Figure 3 and summarized in Table 1.
RNA integrity and profile can be assessed after RNA extraction. Quality assessment of RNA could be done by bioanalyzer instrument (Figure 3A) and only those samples with RIN value higher than 7.0 are qualified to be used for amplification (Table 1).
The quality and quantity of amplified RNA should be evaluated after amplification. The similarity of amplified RNA profile and appropriate size range are the main factors for quality control (Figure 3B). After the two-round amplification, the amplified RNA fragments are very similar in size, ranging between 200 and 800 bp (Figure 3B) and all aRNAs of similar size range are selected for further fluorescence labeling. Also, a successful amplification of the original total RNA will yield at least more than a thousand fold increase of aRNA (Table 1).
Fluorescence labeling efficiency can be calculated according to degree of labeling ratio. The formula is available in the ULS aRNA labeling kit user guide. The user guide suggests the degree of labeling ratio should be 1.0 – 3.6%, indicating that an average of 1 – 3.6 ULS molecules per 100 nucleotides.
The image files after hybridization and scanning can be viewed from FlexArray. A good quality scanned image after microarray hybridization and washing is shown in Figure 4A. If labeled materials are lower or higher than this range, signals will be too low to detect or high background levels will be detected after scanning (Figure 4B).
In current study, positive signal threshold was set at 7.6 (probe signal higher than 7.6 or background < 7.6 considered to be positive). Approximately 46% of the probe sets representing 20826 positive spots are indicated in red color in bovine Day 7 embryos (Figure 5). After removing unannotated and duplicate gene symbols, only 6,765 unique gene symbols is left for PANTHER analysis.
These 6,765 genes represent the unique RNA transcripts expressed in the Day 7 bovine blastocyst. In order to find the biological process specific to bovine blastocyst, PANTHER Overrepresentation Test is performed. The top 10 Gene Ontology (GO) term IDs with the highest fold enrichment index is selected (Figure 6). In general, these specific GO terms largely represent the active cellular division process such as mitosis and chromosome segregation, regulation of cell cycle and initiation of cytoplasmic and mitochondrial translation.
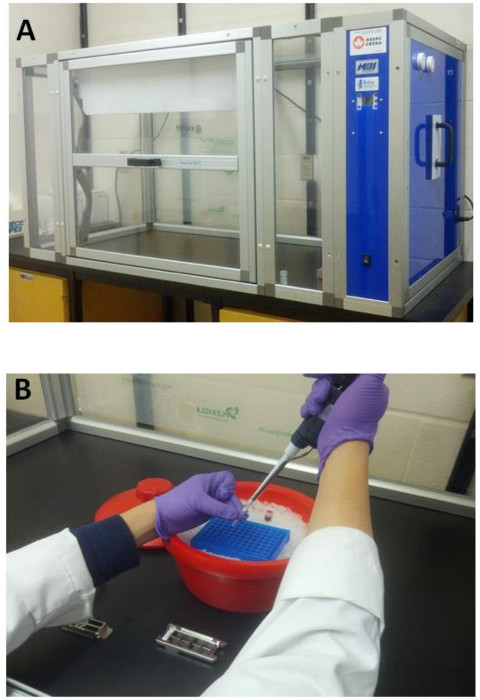
Figure 1: Ozone-free Box (A) and Array Labeling Reaction (B). A) Ozone Free Box consists of a catalytic converter unit which recycles the air and destroys ozone and a highly sensitive ozone sensor to monitor the ozone level inside the box during microarray performance. B) Labeling procedure and array hybridization perform inside the box to avoid the fluorescent Cyanine 647 dye degradation by ozone. Please click here to view a larger version of this figure.
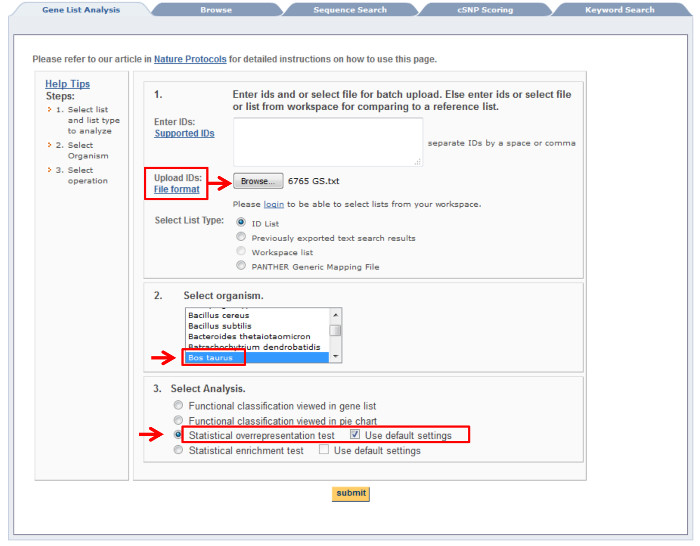
Figure 2: PANTHER Gene List Analysis. Red arrows indicate the area need to be filled. Please click here to view a larger version of this figure.
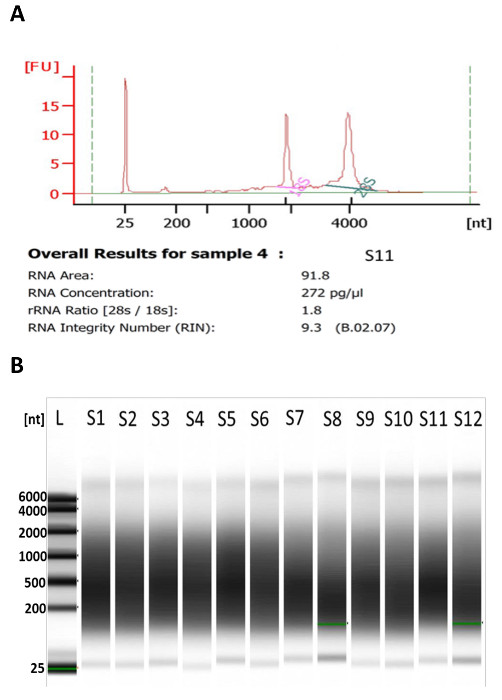
Figure 3: A Representative Bioanalyzer Electropherogram of Total RNA (A) and the Gel Image of Amplified RNA (B). A) Showing overall result with RIN value, RNA concentration and rRNA ratio. B) The Profile of amplified RNA after two-round amplification. Amplified RNA fragments are expected to be in a range between 200 and 800 bp. Please click here to view a larger version of this figure.
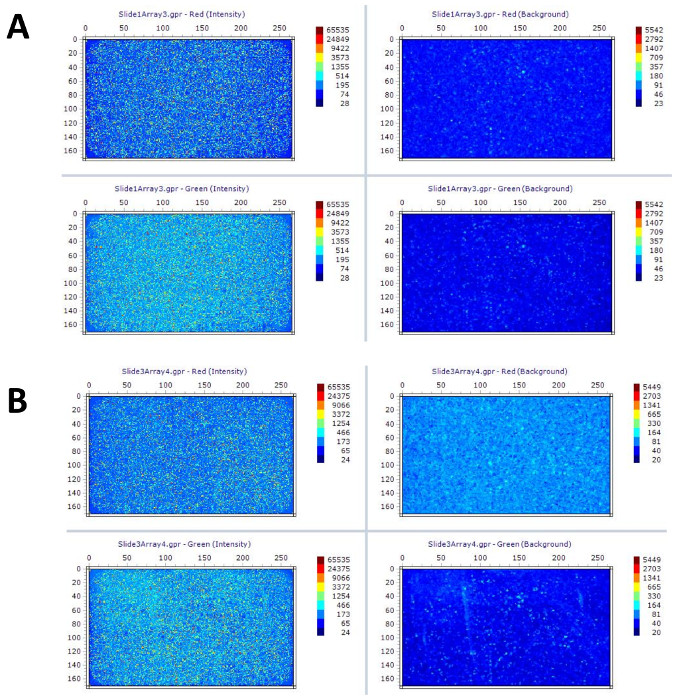
Figure 4: Image of Array Intensity Map. (A) A good image of an array slide showing green and red channel correspond to Cyanine 547 and Cyanine 647 signals after scanning with high spot intensity (indicated on the left with blue green color) and low background (indicated on the right with dark blue color). (B) A bad image of an array slide showing very high background image from red channel (indicated from the upper panel with the similar blue green color from the image of Cyanine 647 spot intensity). Color chart indicates the intensity of signal with numerical values corresponding to different colors. The value of signal intensity is the lowest in the range of dark blue color. Please click here to view a larger version of this figure.
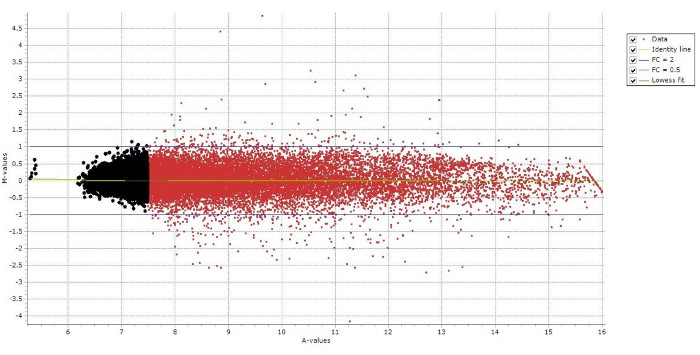
Figure 5: MA Plot for Day 7 Bovine Embryo Gene Expression Profile. 20,826 red spots represented positive signals above background signals. Background spots are highlighted in black color. Please click here to view a larger version of this figure.
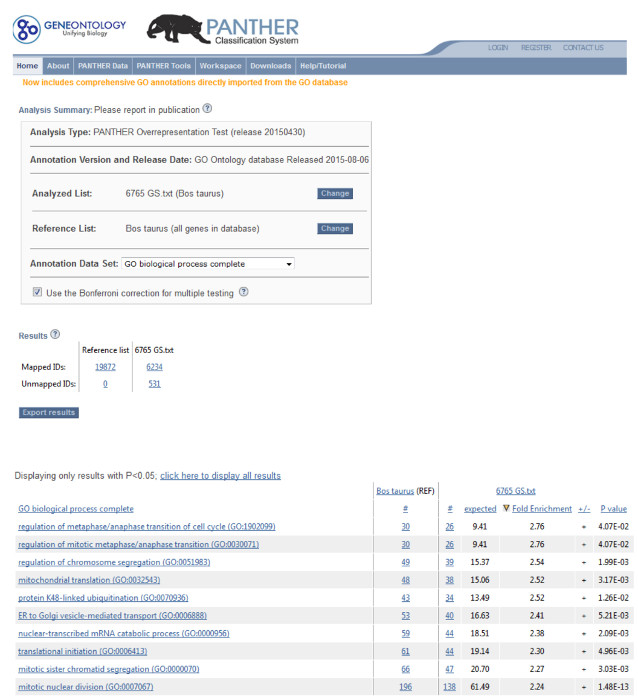
Figure 6: The Top 10 Gene Ontology (GO) Term IDs with the Highest Fold Enrichment Index. These specific GO terms largely represent the active cellular division process such as mitosis and chromosome segregation, regulation of cell cycle and initiation of cytoplasmic and mitochondrial translation. Please click here to view a larger version of this figure.
| Sample | Number of embryo | Tipo | Embryo morphological quality | RNA concentration (pg/µL) | RIN | Used total RNA for amplification (pg) | Amplified RNA concentration (ng/µL) |
| S1 | 4 | Blastocyst | Grade 1&2 | 101 | 8.6 | 858.5 | 3498.4 |
| S2 | 1 | Blastocyst | Grade 2 | 142 | 7.4 | 1207 | 1682.4 |
| S3 | 2 | Blastocyst | Grade 1 | 135 | 8.8 | 1147.5 | 3185.3 |
| S4 | 3 | Blastocyst | Grade 1 | 177 | 9 | 1504.5 | 2906.4 |
| S5 | 5 | Blastocyst | Grade 1 | 636 | 8.3 | 5406 | 3437.5 |
| S6 | 3 | Blastocyst | Grade 1&2 | 159 | 9.1 | 1351.5 | 1689.8 |
| S7 | 3 | Blastocyst | Grade 1 | 154 | 9 | 1309 | 4507.1 |
| S8 | 4 | Blastocyst | Grade 1 | 304 | 8.5 | 2584 | 3089.3 |
| S9 | 5 | Blastocyst | Grade 1 | 282 | 9.1 | 2397 | 3917.8 |
| S10 | 6 | Blastocyst | Grade 1&2 | 374 | 9.4 | 3179 | 2979 |
| S11 | 5 | Blastocyst | Grade 1 | 272 | 9.3 | 2312 | 2576 |
| S12 | 3 | Blastocyst | Grade 1 | 206 | 9 | 1751 | 2567.9 |
Table 1: Sample and RNA information. The concentration and quality of RNA should be evaluated after extraction. RNA integrity and concentration can be assessed by any bioanalyzer. RNA integrity number should be more than 7.0. The RNA concentration should be examined by spectrophotometer instrument after amplification. Embryo quality was evaluated according to the Manual of the International Embryo Transfer Society (3rd ed., Savoy, IL).
