Transcriptomics
To analyze differentially expressed genes by E2 treatment, we chose to perform an RNA-Seq experiment. In addition to providing information about mRNA levels, RNA-Seq data can also be used to monitor changes in non-coding RNA (long non-coding RNAs, microRNAs) and alternative splicing events. We did not provide information on the analysis of non-coding RNAs or alternatively transcribed genes, since scope of our study is to identify protein coding genes that are important for metabolic regulation. However, RNA-Seq is an excellent way of obtaining information on differential gene expression events.
In order to obtain high quality reads, it is important to get high purity RNA. After isolating RNA we purified the RNA using a RNA clean up kit. Unfortunately nearly 50% of the RNA is lost during this process and amount of starting RNA should be taken into consideration to obtain required amount (100-500 ng) by the end of purification step.
We quantified the RNA yields using fluorometer RNA HS assay, which is very accurate and is preferred method for quantification of low-abundance RNA. Next, the integrity and overall quality of the RNA samples were examined using bioanlayzer, which is a viable alternative to gel electrophoresis using a minimal amount of RNA. The analysis shows all the samples have sharp and clean bands of 18S and 28S rRNAs indicating the integrity and purity of the samples (Figure 2B). RNA Integrity Number (RIN) was used as a standard for RNA quality control. RIN 10 indicates intact RNA, RIN 6 partially degraded and RIN3 strongly degraded16.
It is recommended that the RIN to be at least 7 for sequencing experiment. All of our RNA samples received RIN value between 9.6-9.9 (Figure 2A). Sequencing libraries were constructed from 500ng of high-quality RNA from each sample using ultra-high-throughput sequencing system such as Illumina 2500. On average 18,000,000 High-quality sequencing reads were obtained for each sample (Table 1), which was ready for downstream analysis. To check the overall quality of the high-throughput sequence raw data, FastQC was run for each sample's reads. A representative FastQC report for one vehicle RNA-Seq data was shown (Figure 3). The sequence length of the most reads was about 100 bp (Figure 3A). For each read, the quality score was 32-34 for the first 5 bp and 36-38 from 6-100bp (Figure 3B). Among 17,990,863 reads generated from this sample, most of them had quality score higher than 34 (Figure 3C). The GC content per read in the sequence also followed the normal distribution with an average of 48% GC. Overall the quality score of the reads was very high.
Cistromic analysis of ERα binding sites
To achieve the efficient deep sequencing, obtaining DNA fragments at desirable size is one of the important factors. Current sequencing approaches may have different requirements on the DNA fragments, for example 300-600 bp for ultra-high-throughput sequencing system, 100-200 bp for AB/SOLiD17. In this experiment most of the DNA fragments are between 300 bp and 600 bp as shown on gel (Figure 4). As an alternative, readers might chose to prepare their chromatin using MNase digestion, however we feel our established protocol provides the required samples with high-quality and provides reproducible results. Ten ng of DNA from each treatment and its corresponding input sample was prepared in EB buffer. The ChIP DNA was single-read sequenced using ultra-high-throughput sequencing system. ChIP DNA samples yielded about 20,000,000 reads while the input DNA yielded more than 35.000,000 reads (Table 1).
Metabolomics
To quantify the metabolites in mammalian cells using GCMS, at least 10 µg of cell mass are required. We started with 400,000 MCF7 cells with the experiments. By the time of harvest, there were about 500,000 cells in each plate. The overview of the metabolites represented by peaks were compared between the control and the Estradiol treated sample (Figure 5). About 100 metabolites were identified, which count for 60% of the total known in mammalians. Here we present a list of the top 10 metabolites, which show significant changes in E2 treatment (Table 2). Overall, E2 upregulated pathways including fructose and mannose metabolism, histidine metabolism, purine metabolism, cholesterol biosynthesis and vitamin B3 metabolism. The downregulated pathways were butanoate metabolism, de novo fatty acid biosynthesis, pentose phosphate pathway, urea cycle and metabolism of arginine (Table 3).
Data analysis and integration
The data from RNA-Seq, ChIP-Seq and GC-MS were processed and analyzed through a series of steps (Figure 6). After the comparison between genes identified in RNA-Seq and ChIP-Seq, one set of up-regulated and one set of down-regulated genes by Estradiol were extracted. From the metabolic data, we obtained two sets of up- and down-regulated compounds by the Estradiol treatment. Next we used Metscape, an app for Cytoscape, to visualize and interpret the gene expression and metabolic data in the context of human metabolism15. First we chose to include elements of compound, reaction, enzyme, and gene in the network. We selected the compounds/genes as query and two networks of the E2 up-regulation (Figure 7A) and down-regulation (Figure 7B) were shown. The Metscape also provides an option to build networks on selective pathways. For example, using the same set of data as building the down-regulated overall network, a subnetwork of androgen-estrogen biosynthesis and metabolism pathway was generated by switching the query from the compound/gene to specific pathway. The genes or compounds with significant changes were highlighted in brighter color. Besides selecting a specific pathway from the drop-down window as described, one can also create a sub-network by applying "create subnetwork" function to the chosen area. As shown in Figure 6A and B, ERα activation and direct binding to gene regulatory regions impacted many metabolic pathways in breast cancer cells. Our integrative analysis indicated that, ERα directly binds to and regulates expression of FASN gene, which is important for de novo fatty acid biosynthesis pathway (Figure 7C). In addition E2 treatment downregulated androgen-estrogen biosynthesis pathways by repressing expression of CYP4Z1, CYP2E1, CYP4B1, CYP2C8, TPO, GSTA4 and GSTM2 in these pathways.
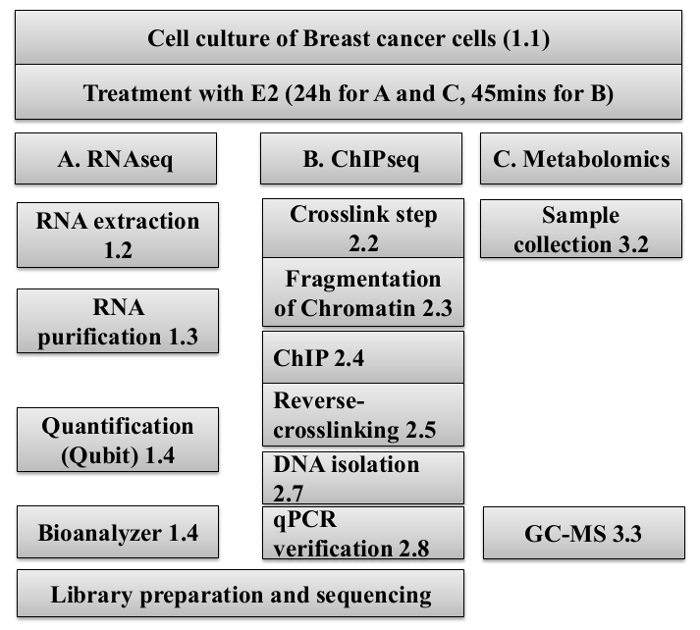
Figure 1: Workflow of the preparation of samples of RNA, DNA and cell lysates for the RNA-Seq, ChIP-Seq, and Metabolomic analysis.
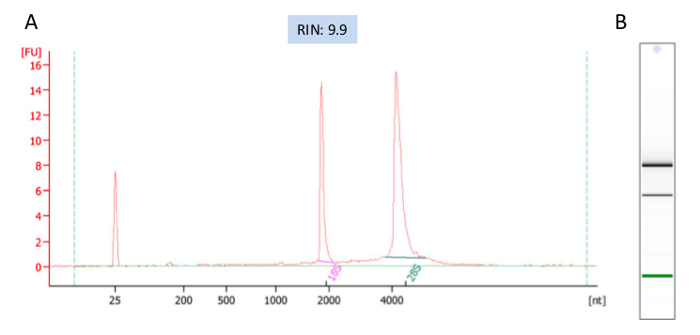
Figure 2: A representative electropherogram of the RNAs analyzed with Agilent 2100 Bioanalyzer. RNA Integrity Number (RIN), indicator of RNA quality, was given by the analysis as 9.9. (A) On a virtual gel picture of the RNA sample both bands of 26S and 18S are sharp. (B) Fluorescent signals of individual trace in the RNA shows that the intensity of 28S peak is greater than 18S peak and no contamination is seen. Please click here to view a larger version of this figure.
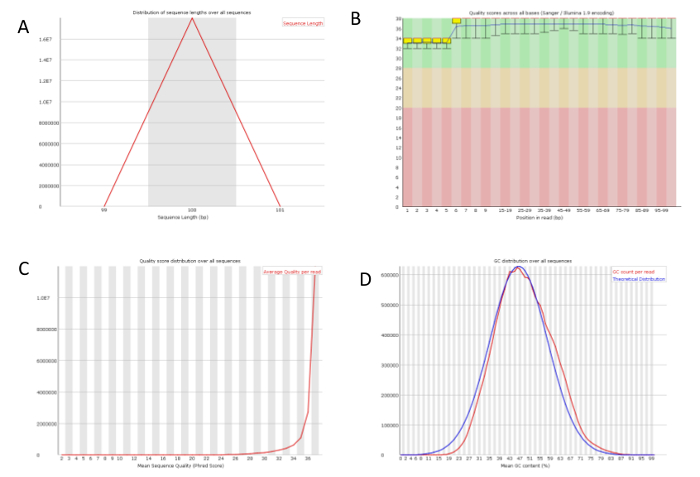
Figure 3: A representative quality check on the RNA-Seq raw data. One of the vehicle sample's sequence data was analyzed using FastQC. (A) The sequence length distribution of the library which is 100 bp. (B) The quality score along the position in read. (C) The quality score per sequence showed the universal high quality of all sequences. (D) The GC content per read matched the theoretical distribution. Please click here to view a larger version of this figure.
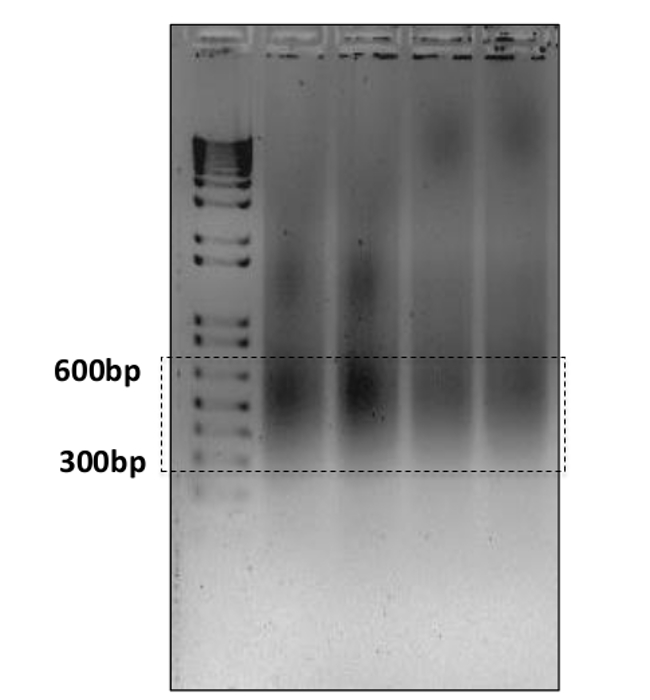
Figure 4: Gel electrophoresis of the sonicated chromatin fragments. 5 µl of the cell lysate from each sample was run on a 1.5% agarose gel with a 1,000 bp plus ladder on the first lane. The majority of the fragments are between 300 and 600 bp. Please click here to view a larger version of this figure.
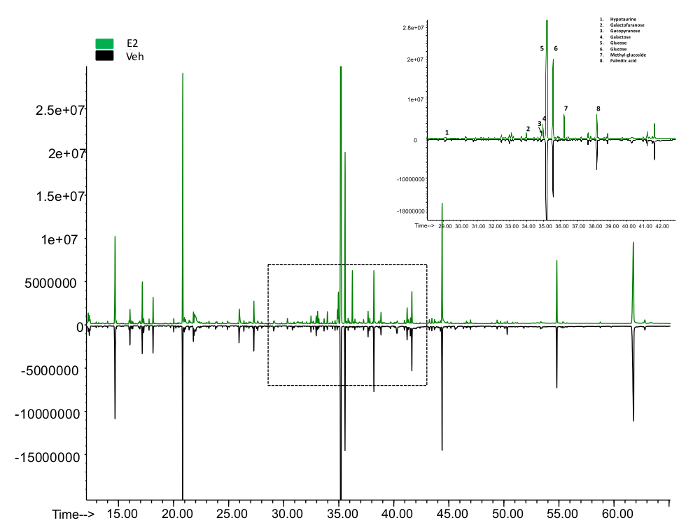
Figure 5: The overview of peaks representing metabolites from the GC-MS. The upper panel shows the peaks identified in the E2 treated samples while the lower panel from the control. The area enclosed was roomed in to show the exact metabolite represented by each peak. Please click here to view a larger version of this figure.
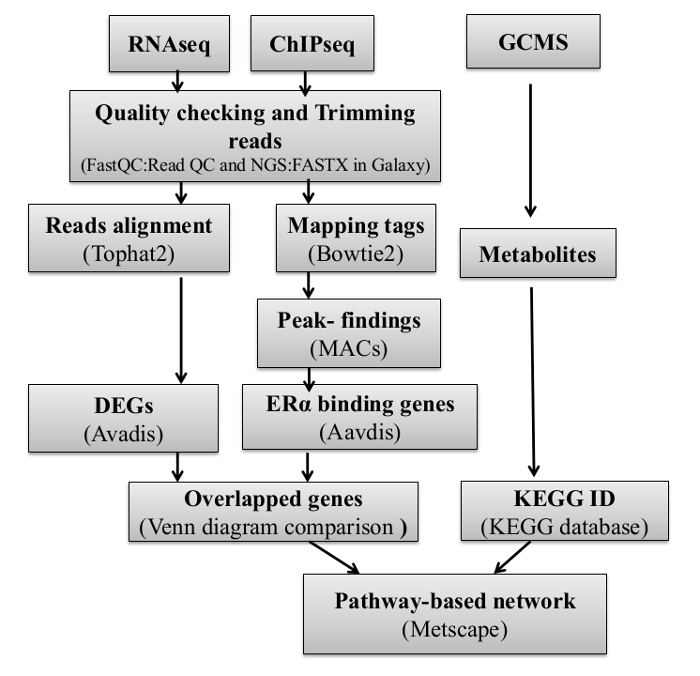
Figure 6: Flowchart of the Integrative clustering method.
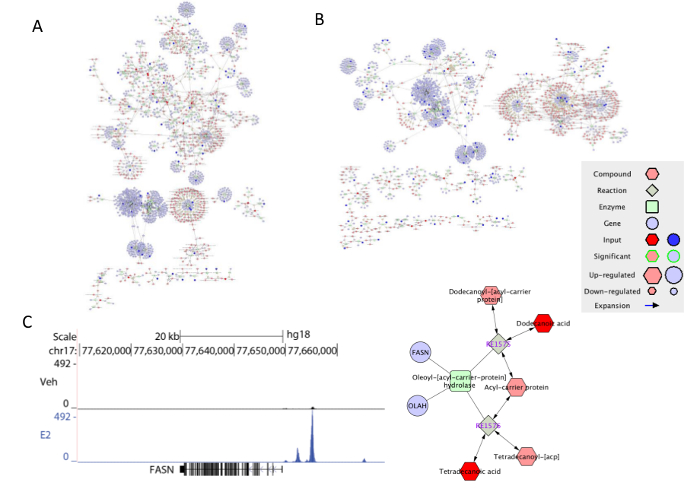
Figure 7: The visualization of the integrated gene-metabolite data in Metscape. (A) The network of pathways up-regulated by E2. (B) The network of pathways down-regulated by E2. (C) E2 stimulated ERα recruitment to the nearby region of FASN, which is found to be regulated in the de novo fatty acid biosynthesis network generated by applying subnetwork function. Please click here to view a larger version of this figure.
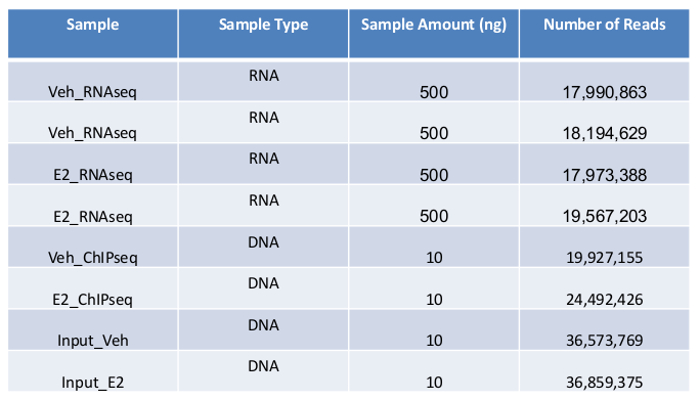
Table 1: Summary of ultra-high-throughput sequencing of RNA-Seq and ChIP-Seq.
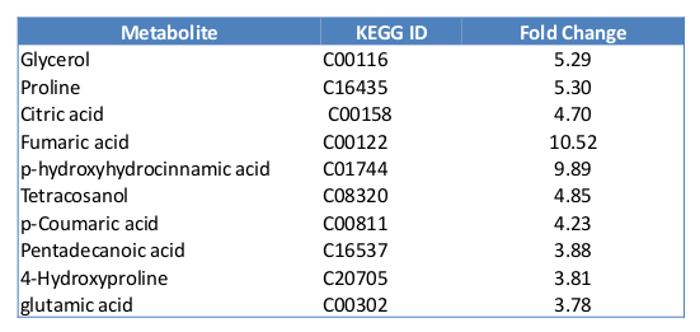
Table 2: List of top 10 metabolites with significant changes with E2 treatment.
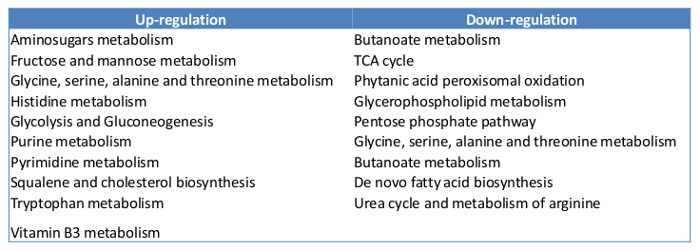
Table 3: Pathways up- and down-regulated by E2 treatment.
