Isolation of High Density Lipoprotein After Removal of Exosomes
To obtain miRNA from highly purified HDL it is necessary to remove exosomes that represent a source of miRNA contamination7. This was done prior to density gradient ultracentrifugation with a commercially available kit. For practical purposes a three step standard density gradient ultracentrifugation protocol developed by commercial company was modified (Figure 1). This protocol requires centrifugation with a fixed-angle rotor at a speed of 140,000 rpm which is substantially faster than commonly used protocols with centrifugation forces up to 54,000 rpm3, 8, 9, and 10. Employing a widely available T-1270 rotor with a maximum force of 70,000 rpm, centrifugation was initially carried out with polyallomer tubes which have failed to resist the centrifugation forces. To avoid collapse of tubes, polycarbonate tubes were successfully used. Centrifugation time is critical for lipoprotein oxidation and potentially miRNA degradation11. Therefore several different centrifugation times, ranging from a total of 8 to 96 hr were tested. Furthermore temperature at which centrifugation was carried out was adjusted based on centrifugation time and force, respectively.
Purity of the High Density Lipoprotein Fractions
Purity of isolated high density lipoprotein fractions were checked with agarose gel electrophoresis. Figure 2. illustrates a typical electrophoresis result for the HDL subtraction isolated by gradient ultracentrifugation. It clearly showed that HDL was devoid of any contamination of VLDL and exhibits typical α-mobility. The absence of any α-migrating lipoproteins in the VLDL and LDL fractions demonstrates the complete recovery from HDL during the ultracentrifugation step. The HDL fraction, however, also showed some trace of β-mobility, a known electrophoretic banding pattern due to contamination with lipoproteins (Lp(a))12.
Elimination of Lp(a) from Isolated HDL
Studying miRNAs carried in HDL requires isolation of HDL of highest purity. Interference of Lp(a) with HDL potentially contributes to cross-contamination of HDL with miRNAs carried in LDL. To minimize this contamination of HDL, 15 μl of β-mercaptoethanol 10 was added to solution C during the last centrifugation step (Figure 1). Addition of β-mercaptoethanol has been shown not to change HDL density properties10. As displayed in Figure 3., addition of β-mercaptoethanol resulted in the absence of any b-migrating lipoproteins consistent with very effective removal of Lp (a).
Purification of miRNA from HDL
Isolation of miRNA was initially attempted employing lysis reagent which is commonly used for RNA extraction from blood. Although this method resulted in a very good RNA yield (91.45 ng/µl) but after spectrophotometric analysis showed unsatisfactory RNA purity. Additional purification of the isolated RNA by removing phenol improved the purity but the RNA yield was now significantly low. Next, another kit was tested which showed acceptable RNA purity (260/280 nm ratio of 1.7) but the RNA yield was 26-fold lower compared with the lysis reagent (3.45 vs. 91.45 ng/µl). The best results for acceptable RNA purity with a good yield were obtained with the miRNeasy serum/plasma kit (48.2 ng/µl; 260/280 nm ratio of 1.6) and therefore this extraction procedure was subsequently employed for the detection of miRNA from the isolated HDL lipoprotein fraction.
To demonstrate the feasibility to quantify miRNAs carried in human HDL purified with this method, we chose to amplify miR-223. This miRNA was chosen because miR-223 was identified in cargo of HDL3 and was shown to repress HDL cholesterol uptake5. The Figure 4A. shows a typical log plot of amplification curves comparing baseline threshold and threshold cycle values after optimization of the PCR reaction for the miR-223 and the spiked-in Cel-mir-39. This synthetic gene was used as internal control as there are no reports of known normalization control for miRNA in plasma. In addition, several potential established endogenous housekeeping genes like RNU6-2, RNU-48, HY3 could not be detected and SNORD95 could be detected in purified HDL, but, the difference in Ct values of miR-223 and SNORD95 is less than five (data not shown). Melting curve analysis illustrated in Figure 5. clearly shows a distinct single peak consistent with amplification of a selective miRNA in the preceding PCR. As illustrated in Figure 4B., both miR-223 and the reference Cel-mir-39 could consistently be detected in all six pro bands. Relatively small variations among all individuals were observed for Cel-mir-39 compared with miR-223. These findings support isolation of HDL at high purity to allow detection of its miRNA cargo.
Detection of miR-223 in Purified HDL
Real time quantitative PCR method was used to quantify double hairpin-structure miRNA precursors to single strand miRNA. This method needs a forward/reverse gene-specific primers and a thermostable reverse transcriptase to convert the hairpin structure of the miRNA to cDNA. The cDNA was subsequently amplified and quantified using real-time qPCR with the help of SYBR green detection. MicroRNA from serum, plasma and purified HDL plasma can be accurately profiled using the miRNA RT PCR system.
Our experimental product of mean miR-223 gene expression showed a Ct value of 30.9 in purified HDL and its corresponding negative controls were above cut-off 35 (NTC = 38.1 Ct value, NRT and NAC were not amplified). In this experiment, we saw in the purified HDL samples the formation of Primer-dimer with low melting temperature (73 °C in miR-223 and 75.5 °C in Cel-miR-39) and higher Ct value than the specific miRNA assays (Table 1). The Primer-dimers in Table 1. occur probably because of high concentration of primers in the solution. This occurs mostly when primer molecules attach to each other after 30 PCR cycles 13. This allows them to anneal to other primer molecules and in effect, become linear mini-templates and it will cause higher background and may lead to a generation of Ct value < 40 for NTC (No template control) samples.
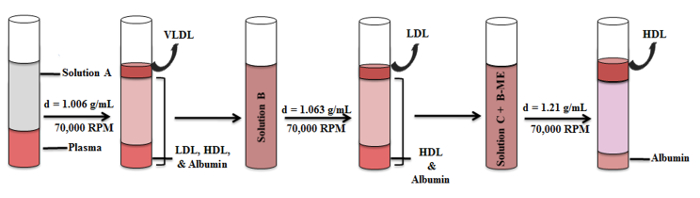
Figure 1. Schematic Representation of the HDL Isolation Procedure. HDL was prepared by density gradient ultracentrifugation in a series of three centrifugation steps. Distribution of lipoprotein bands and intermediate fractions in the density gradient are illustrated. Please click here to view a larger version of this figure.
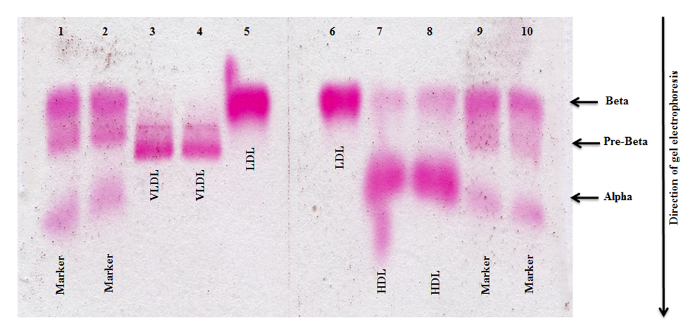
Figure 2. Agarose Gel Electrophoresis of Lipoproteins Isolated Without β-mercaptoethanol. VLDL, LDL and HDL fractions isolated by density gradient ultracentrifugation without adding β-mercaptoethanol to solution C demonstrate the presence of Lp (a) in the HDL fraction. Lanes 1, 2, 9, and 10 each represent the size marker; Lanes 3/4, 5/6 and 7/8 represent VLDL, LDL and HDL respectively. Please click here to view a larger version of this figure.
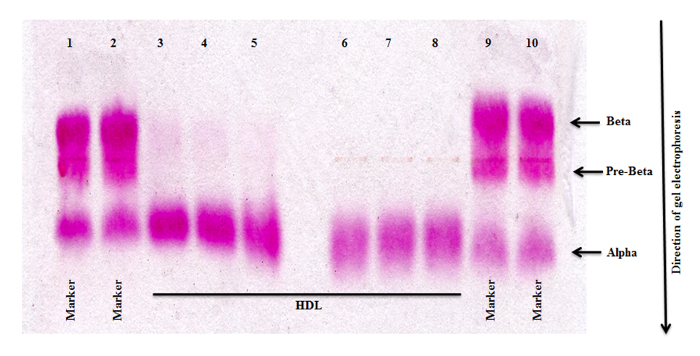
Figure 3. Agarose Gel Electrophoresis of HDL Isolated with β-mercaptoethanol. Adding β-mercaptoethanol to solution C before the last centrifugation step resulted in the removal of Lp(a) from the HDL fraction. Lanes 1, 2, 9 and 10 each represent the size marker; Lanes 3-8 represent HDL. Please click here to view a larger version of this figure.
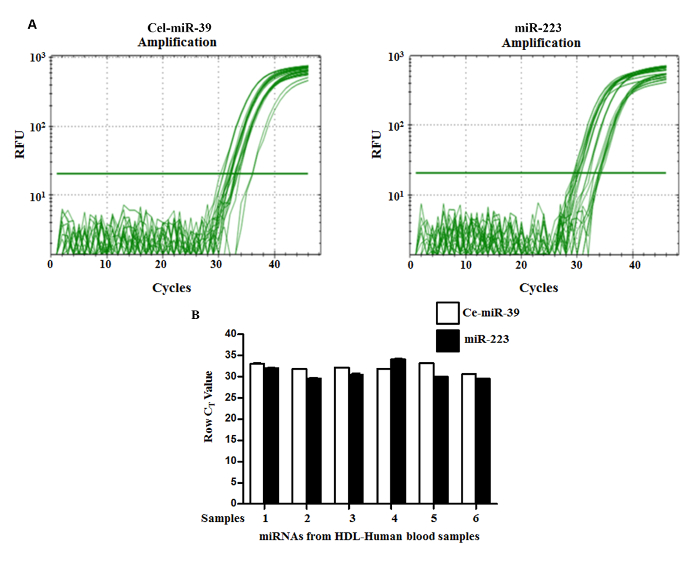
Figure 4. Amplification Plot of Two Different miRNAs and Expression of Cel-miR-39 and miR-223. (A) Real-time quantitative PCR employing isolated HDL was carried out as described. The PCR was run for 45 cycles and the point at which the curve intersects the threshold is the CT-Value. The data show the expression of Cel-miR-39 (left panel) and miR-223 (right panel), respectively. The horizontal line represents the detection threshold. A cycle number of 35 was set as cutoff for positive amplification. (B) Representative real-time PCR data from isolated HDL obtained from 6 samples are shown. Raw mean Ct values are shown for Cel-miR-39 and miR-223, whose expression levels vary less than 2 Ct value in purified HDL plasma fraction between 6 samples, each from a different donor. Expression profiling was performed using the PCR System and the Human Serum & Plasma miRNA qRT-PCR. Please click here to view a larger version of this figure.
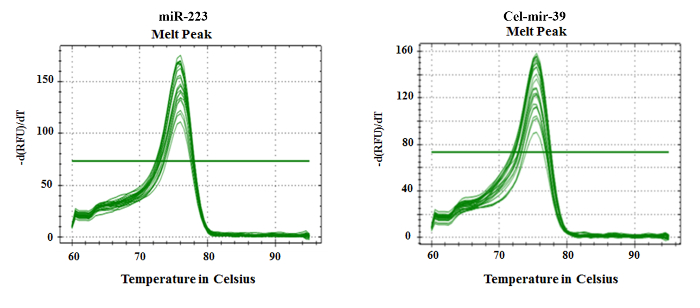
Figure 5. Melting Curves for Cel-miR-39 and miR-223. As illustrated, the presence of a single peak indicates specific amplification of Cel-miR-39 (right panel) and miR-223 (left panel), respectively. Please click here to view a larger version of this figure.
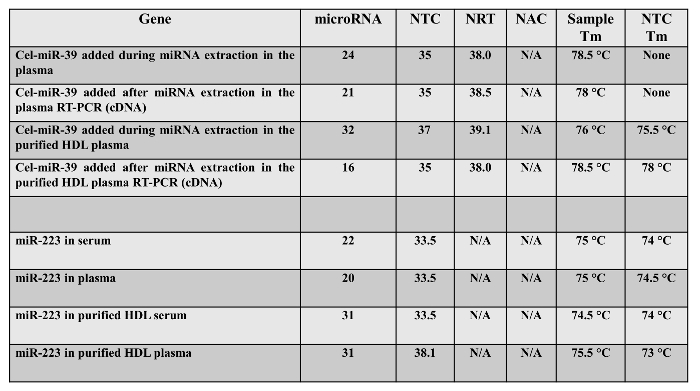
Table 1. Row Ct value of negative and positive control of synthetic Cel-miR-39 and miR-223 characterized in serum, plasma, cDNA, and purified HDL fraction. NTC (No template control), NRT (No reverse transcription enzyme), NAC (No amplification control, only water and reagents of qRT-PCR).
