Microinjection for gene regulatory network studies
Embryos of the ascidian Ciona intestinalis are well suited for gene functional or gene regulatory studies at cell lineage level and with cellular resolution. mRNAs, MOs or labels can be microinjected into the unfertilized egg or into individual blastomeres following fertilization. MO mediated gene knockdown using the microinjection technique is described in step 6. MOs specifically target the mRNA of selected genes and prevent their translation into protein. MO mediated loss-of-function (LOF) of developmental genes changes the expression of a broad array of downstream genes, overall revealing a glimpse into the embryonic GRN10. Such analyses in Ciona provided the first whole embryo regulatory blueprint in a metazoan11. Figure 1 displays an example of how microinjection was utilized to study the upstream regulation of the expression of the conserved Ci-Myelin Transcription Factor (Ci-MYT) at the gastrula stage of Ciona embryos. Monitored by in situ hybridization (ISH), endogenous expression (Figure 1a, schematized in Figure 1a') is observed in the 6-row neural plate precursors, of notably the brain (rows III/IV, in red) and the spinal cord (row I, in green). In addition, Ci-MYT upstream regulation was analyzed by MO injection targeting several factors that are expressed in or adjacent to neural precursors prior to Ci-MYT expression onset (Figure 1b-g). MO knockdown of early embryonic factors, such as the Forkhead box A-a (FoxA-a) transcription factor and the fibroblast growth factor 9/16/20 (FGF9/16/20) (Figure 1b or 1c, respectively) eliminates overall Ci-MYT expression. Other transcription factors only partially affect Ci-MYT expression resulting in MO-mediated downregulation in a subset of brain precursors (blue arrow heads in Figure 1d, f and g). Conversely, Ci-MYT expression is ectopically activated upon downregulation of Nodal (Figure 1e, red arrow heads), suggesting that Nodal normally represses Ci-MYT expression in these lateral nerve cord precursors.
Electroporation for efficient gene functional and enhancer studies
Electroporation of plasmid DNA into Ciona zygotes is an efficient method for transient transgenesis and subsequent observation of phenotypic changes in vivo. Unlike microinjection of mRNA, electroporation allows for tissue specific overexpression, in which gene coding regions (ORFs, open reading frames) are expressed under control of tissue specific drivers. These normally constitute cis-regulatory regions of well-known genes (such as the brachyury enhancer for expression in notochord precursors8). Conversely, electroporation is instrumental for the efficient analysis of novel regulatory regions where reporter genes (LacZ or green fluorescent protein, GFP) reveal their activity in vivo. The workflow for the identification and subsequent electroporation mediated analysis of such a novel regulatory region (for Ci-MYT) is shown in Figure 2.
A vast repertoire of Ciona community data, readily accessible in ascidian specific genome browsers12,13, like the Ascidian Network for In situ Expression and Embryological Data (ANISEED)23, is inspected for the in silico identification of regulatory regions (Figure 2A). Subsequent polymerase chain reaction (PCR) based cloning of these regions into expression vectors allows for electroporation mediated testing in vivo. (Figure 2B). The genome browser screen-shot in Figure 2A shows the upstream region of the KH transcript model for Ci-MYT (first three exons, in orange, and two introns are visible). Different genome browser tracks annotate functional genome data predicted for this region, such as chromatin immunoprecipitation (ChIP) data14 (here shown for ZicL, Ci-zinc finger of the cerebellum L), sequence conservation between two related Ciona species15,24 or nucleosome occupancy16,17 (from top to bottom in Figure 2A). Generally, exonic protein coding regions are highly conserved (alignment track in Figure 2A). Additional high conservation peaks appear in non-coding regions upstream and in the first intron. Two of these are predicted to be nucleosome free according to a sequence based algorithm16,17 (red frames in Figure 2A) suggesting accessibility to transcription factors. Indeed, Chip-on-ChIP signals14 for Ci-ZicL transcription factor binding (green bars in Figure 2A) are enriched in these two conserved regions. Furthermore, using an interface25 that searches for potential transcription factor binding sites, we identified ZIC sites located within the genome sequences marked by Ci-ZicL ChIP clusters (orange arrows and sequences with underlined ZIC-core, Figure 2A). The binding of Ci-ZicL transcription factor in conserved and predictably nucleosome free regulatory regions of Ci-MYT is consistent with the downregulation of Ci-MYT expression observed in MO-injection mediated knockdown experiments targeting ZicL (Figure 1d).
After determining potential cis-regulatory regions in silico for Ci-MYT expression, these are PCR amplified from Ciona genomic DNA and inserted by recombination cloning into LacZ reporter plasmids19. The constructs are then electroporated into fertilized Ciona eggs (Figure 2B) and analyzed for their activity by LacZ staining (Figure 3).
Figure 3 shows that by such an in silico approach it was possible to identify two separate cis-regulatory sequences that recapitulate Ci-MYT mRNA expression domains (compare Figure 1a). Generally, transiently transgenic embryos electroporated with plasmid DNA show mosaic expression in those cells only that have incorporated the DNA. Mosaic LacZ expression is thus driven by pmyt-R3 and can be found in all the precursors that also express Ci-MYT mRNA i.e. neural plate precursors of the posterior row I and the anterior rows III/IV at gastrula and early neurula stages (Figure 3a, b, purple and red arrow heads, respectively). In contrast, pmyt-R5 is active only in posterior (row I) neural plate precursors and starting at the later neurula stage (Figure 3c, red arrow heads). The two regions thus encode two separate spatial and temporal aspects of Ci-myt gene regulation. Interestingly, both regulatory regions also stain additional precursors not seen in the ISH, notably in the anterior and lateral neural plate and in muscle (Figure 3a, b, pink, white, and yellow arrow heads, respectively). Such wider expression may point to repressive elements that are not contained in the isolated regulatory fragments (such as for Nodal that represses lateral neural fate, see Figure 1e) or to low expression levels detected with accumulating LacZ enzymatic signal.
In addition to enhancer studies, electroporation in Ciona also facilitates the functional analyses of Ciona coding genes by their overexpression. A large collection of Ciona full length ORFs is available to the community, and was furthermore annotated for human orthologs, including disease-associated genes18. Individual genes or groups of genes from this recombination-cloning compatible full ORF cDNA library (Figure 2B) are easily transferred into destination vectors containing appropriate drivers to be expressed in embryos. Resulting expression clones can furthermore be co-electroporated with cis-regulatory reporter constructs to test their putative role in enhancer activation. Figure 2B illustrates the isolation of full ORF clones for the transcription factors Ci-ZicL and Ci-GATAa and their recombination into adapted destination vectors that may contain translational tags19 (e.g. green fluorescent Venus- or viral hemagglutinin (HA)-tag) and a tissue specific driver such as the Friend Of Gata regulatory region (pfog driver) for early pan-ectodermal expression15 used in the present study. The effect of Ci-ZicL overexpression in ectodermal and neuroectodermal tissues was analyzed by co-electroporation of pmyt-R3>LacZ and pmyt-R5>LacZ reporters (Figure 3b', b'' and c', c'', respectively). Our electroporation analyses suggest that Ci-ZicL may activate Ci-MYT expression through pmyt-R3 and pmyt-R5 enhancer regions as both reporter constructs showed ectopic LacZ expression in ectodermal regions (green arrow heads in Figure 3b', b'' and c', c'',). As shown in Figure 3d, electroporation of plasmid DNA in hundreds of Ciona eggs allows for statistically relevant quantification of expression changes, illustrated for ectopic pmyt-R3>LacZ expression upon concentration dependent, pan-ectodermal Ci-ZicL expression.
Electroporation for in vivo subcellular localization
Figure 4 depicts the subcellular localization of tagged transcription factors when expressed upon electroporation in ectodermal blastomeres using adapted expression constructs (schematized in Figure 2B). By C-terminally tagging transcription factors (such as Ci-GATAa) with a Venus tag (Figure 4b) or a HA-tag (Figure 4c) and overexpressing them in ectodermal tissues (pfog) we could observe that in vivo,'Ci-GATAa is mostly localized to nuclei, as expected for a transcription factor, whereas Venus expression is ubiquitous, including the cytoplasm. Similar experiments may be performed to study the dynamics of subcellular localization over time, of individual or combinations of transcription factors, possibly revealing their co-localization with different tags.
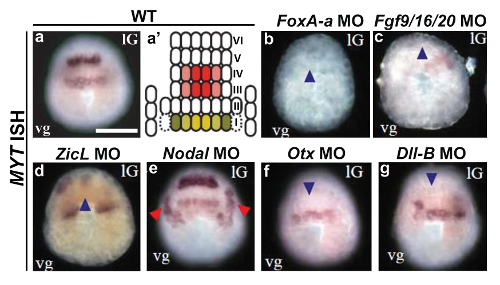
Figure 1: Assessing Ci-MYT regulation by microinjection in Ciona eggs. Ci-MYT mRNA expression in WT and Morpholino (MO) injected embryos. (a) WT expression pattern of Ci-MYT in the neural plate. (a') Schematic representation of stained precursors in the neural plate (NP). Row I/II: posterior neural fate; row III/IV: anterior neural fate; row V/VI: palp fate. (b–g) Ci-MYT expression upon microinjection of various MOs for knocking down indicated genes that participate in the early Ciona GRN (pictures adapted from Imai et al., 2006)11. Ci-MYT, Ci-myelin transcription factor; WT, wild type; blue arrowheads: loss of Ci-MYT signal; red arrowheads: ectopic Ci-MYT signal. Scale bar = 100 µm refers to all images. Please click here to view a larger version of this figure.
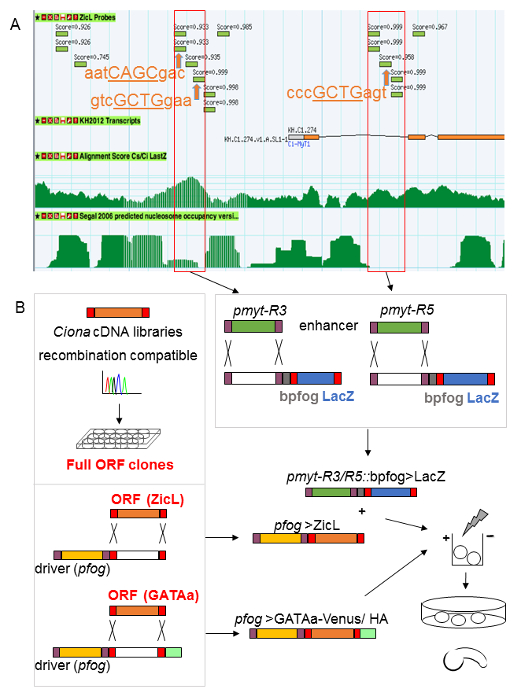
Figure 2: Workflow for in silico search of enhancer regions and recombination cloning for transient transgenesis by electroporation in Ciona embryos. (A) A snapshot from the ANISEED23 ascidian genome browser identifies upstream regulatory sequences of Ci–myt. Red frames mark two potential cis-regulatory regions, pmyt-R3 (scaffold_196:76883..77079) and pmyt-R5 (scaffold_196:75856..76041) that were selected for analysis by electroporation. Names of the tracks: ZicL probes: ChIP-on-Chip data14; KH2012 Transcripts: Ciona intestinalis transcript models; Alignment Score Cs/Ci LastZ: sequence conservation between Ciona savignyi (Cs)/Ciona intestinalis (Ci)15,24; Segal 2006 predicted nucleosome occupancy version 1: chick trained computational algorithm by Segal et al., 200616 applied to Ci as in Khoueiry et al., 201017. Green bars: Ci-ZicL ChIP peaks14; orange arrows: predicted ZIC sites; GCTG: core ZIC binding site. (B) Scheme for generating overexpression constructs from a Ciona full length ORF cDNA library, recombined into destination vectors containing tissue specific drivers (pfog) and protein tags subsequently co-electroporated with reporter constructs (such as pmyt-R3>LacZ or pmyt-R5>LacZ) for in vivo readout. Ci-MYT, Ci-myelin transcription factor; Ci-ZicL, Ci-Zinc finger of the cerebellum L transcription factor; pfog, regulatory region of Ci-Friend of GATA. Please click here to view a larger version of this figure.
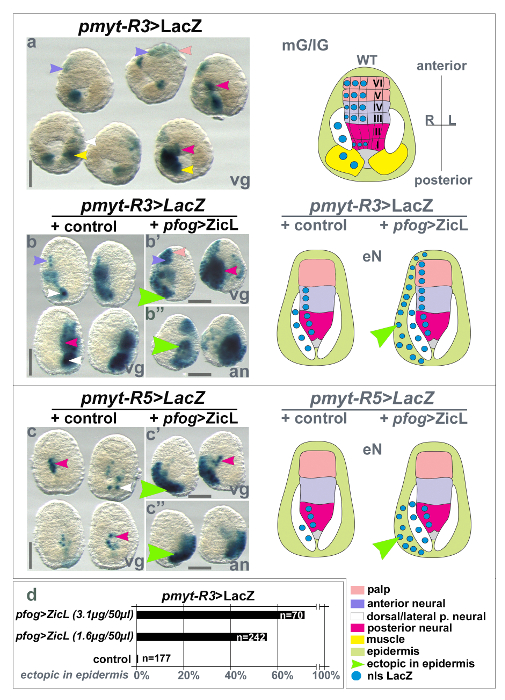
Figure 3: Electroporated Ciona embryos showing Ci-ZicL inducible spatio-temporal cis-regulation of Ci-MYT. LacZ stained embryos at mid-gastrula (mG)/late-gastrula (lG) or early neurula (eN) stage are shown that were co-electroporated with either pmyt-R3>LacZ or pmyt-R5>LacZ and a control construct (pfog>mCherry) or pfog>ZicL. The pfog driver19 mediates ectopic (pan-animal) ZicL expression in ectoderm and neuroectoderm and causes ectopic LacZ stain in these cells. (a) pmyt-R3 LacZ activity in mG/lG stage embryos (small arrowheads, left panel) or in correspondingly colored schematic territories (blue circles, right panel); vegetal view (vg). Row I/II: posterior neural fate; Row III/IV: anterior neural fate; Row V/VI: palp fate. (b) pmyt-R3 activity at eN stages in tissues as in a. (b') Ectopic pmyt-R3 activity upon Ci-ZicL co-electroporation is indicated by green arrowheads. (b'') Same embryos as in b', oriented animal pole up (an). (c) pmyt-R5 LacZ activity at eN stages (c') Ectopic pmyt-R5 activity upon Ci-ZicL co-electroporation is indicated by green arrowheads. (c'') Same embryos as in c' but oriented animal pole up (an). (d) Quantification of representative experiments for Ci-ZicL over-activating pmyt-R3 at low concentrations. One biological repeat is represented. Ci-MYT, Ci-myelin transcription factor; Ci-ZicL, Ci-Zinc finger of the cerebellum L transcription factor; Ci-FOG, Ci-Friend of GATA; WT, wild type; mG, mid-gastrula; lG, late-gastrula; eN, early neurula; an, animal view; vg, vegetal view; nls LacZ, nuclear localization signal for LacZ. Scale bar = 100 µm. Please click here to view a larger version of this figure.

Figure 4: Differential subcellular localization of proteins upon electroporation. Localization of Venus-tagged or Hemagglutinin- (HA)-tagged proteins overexpressed in ectodermal cells using the pfog driver19. (a–c) DIC images. (a', b') Corresponding fluorescence images. (c') Corresponding fluorescence image upon immune histological labelling with TRITC. Scale bar = 100 µm refers to all images. DIC, Differential Interference Contrast. Please click here to view a larger version of this figure.
