Microscope-based method to monitor the different stages of phagocytosis is presented. The different events during the phagocytosis of various fluorescent particulates by DC2.4 cells are shown. Using the techniques described here, we investigated the role of sphingolipids in the early stages of phagocytosis. For this purpose, DC2.4 dendritic cells genetically deficient in Sptlc2, the enzyme that catalyzes the first and rate-limiting step in the sphingolipid biosynthetic pathway, were used. As compared to wild type cells, Sptlc2-/- DC2.4 cells have significantly reduced level of sphingolipids including ceramide, sphingomyelin and glucosylceramide8. Sptlc2-/- DC2.4 cells are defective in binding, as well as in the uptake of C. albicans, zymosan and IgG latex beads8. In Figure 1, the binding of fluorescently conjugated-zymosan particles in DC2.4 cells is shown (see Section 3.1 for details). Sptlc2-/- DC2.4 cells showed significantly less binding of zymosan than the control DC2.4 cells (p = 0.0008; Figure 1A). The number of zymosan particles bound per cell was significantly higher for control cells than for Sptlc2-/- DC2.4 cells (p < 0.0001; Figure 1C). We next investigated the ability of Sptlc2-/- DC2.4 cells to phagocytose C. albicans. Sptlc2-/- DC2.4 cells showed significantly less phagocytosis of C. albicans (p < 0.0005) as compared to the control cells (Figure 2A, 2B). As expected, Sptlc2-deficient cells showed significantly higher number of non-phagocytic cells (p < 0.05; Figure 1C) and decreased number of C. albicans per cell (Figure 1C, D). These results underscore the role of an intact sphingolipid biosynthetic pathway in binding, as well as in the uptake of particulates. Figure 3 shows the time-lapse images demonstrating the early stages of phagocytosis (see Section 3.3 for detailed procedure). Movie 1 shows the live imaging of DC2.4 cells stably expressing F-actin, a biosensor that reveals the distribution of filamentous actin in living cells (see Section 3.1 for more details).
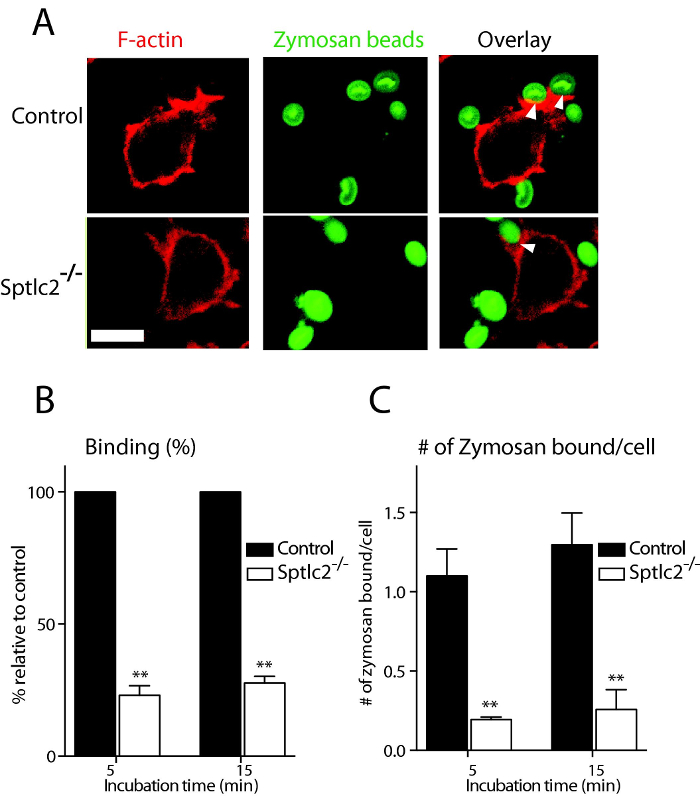
Figure 1: Binding assay of zymosan in DC2.4 cells. (A) Control and Sptlc2-/- DC2.4 cells were incubated with fluorescently conjugated zymosan, and their ability of binding the particulates examined by confocal microscopy. Arrows indicate sites of binding. Scale bar = 100 µm. (B, C) Quantification of the number of bound zymosan particles (B) and the number of zymosan particles bound per cell (C) is shown. Bound particles were quantified and presented as the percentage relative to the control. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
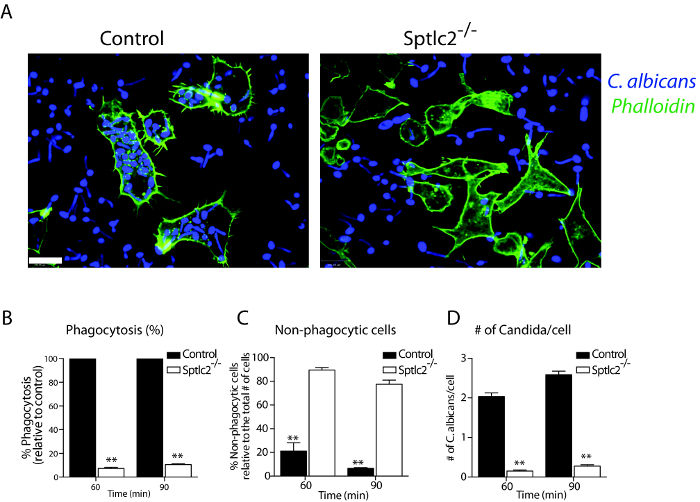
Figure 2: Phagocytosis of C. albicans in DC2.4 cells. (A) Cells were infected with Candida-BFP as described in Step 3.2. At 90 min post infection, cells were fixed and stained with fluorescently conjugated phalloidin and imaged using confocal microscopy. (B–D) Quantification of the number of internalized Candida-BFP, non-phagocytic cells, and the number of Candida-BFP per cell is shown. All graphs display SD of three independent experiments, and at least 200 cells were counted for each experiment. Unpaired t-test was used to analyze the significance of the observed differences. ** p < 0.001. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
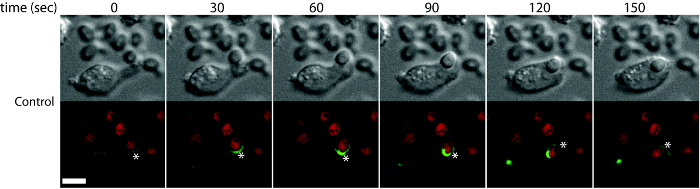
Figure 3: Time-lapse imaging of C. albicans uptake to visualize the early stages of phagocytosis. Wild type DC2.4 cells stably expressing F-actin-mCherry (F-actin) were incubated with Candida-BFP (shown in red) and imaged using confocal microscopy. Images captured at 30 sec intervals are shown. Asterisks show the different stages of actin remodeling during phagocytosis. Scale bar = 100 µm. Reprint with permission from Tafesse et al., 2015 8. Please click here to view a larger version of this figure.
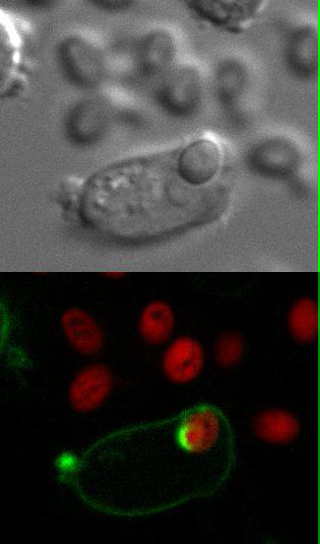
Movie 1: Live imaging. Wild type DC2.4 cells stably expressing F-actin-mCherry (shown in green) were infected with Candida-BFP (shown in red). Live imaging was performed using confocal microscopy as described in Step 3.3. Reprint with permission from Tafesse et al., 2015 8. Please click here to view this video. (Right-click to download.)
