1. Real Time Intracellular Growth and Eukaryotic Cell Cytotoxicity Assay
- Preparing Host Cells (J774A.1 Cells)
- Culture J774A.1 Mus musculus macrophage-like cells in suspension in RPMI 1640 with 9% iron-supplemented calf serum. Initially passage in tissue culture flasks. After cells have become confluent in a 75 cm2 tissue culture flask in 15 ml of medium, split by scraping and dilution to 65 ml with the same type of medium, of which 15 ml is returned to the tissue culture flask and 50 ml is transferred to a 250 ml bacterial shaker flask.
- For scale-up, culture in suspension in 250 ml and/or 1,000 ml bacterial flasks filled to one fifth volume with tissue culture medium. Aerate by setting rotation speed at approximately 120 revolutions per minute. For consistent growth, incubate at exactly 5% CO2 and 37 °C.
- Harvest cells when they reach density in the range 2.5 x 106 cells/ml to 5 x 106 cells/ml. Ensure dead cell percentage (most easily assayed by trypan blue staining) does not exceed 25%, as dead cells will increase background noise in cytotoxicity assays.
- Plate in white, tissue culture-treated, 384-well microplates at 5 x 104 J774A.1 cells in 30 µl tissue culture medium per well. Incubate microplates overnight to achieve 90% confluence on day of experiment.
- Preparing Luminescent or Fluorescent Legionella pneumophila
- Passage L. pneumophila (Lp02::flaA::lux8) bacteria on appropriate medium, i.e., buffered charcoal yeast extract (BCYE) medium. If using a thymidine auxotrophic strain, supplement medium with 100 µg/ml thymidine. Prepare patches of bacteria for experiments by spreading organisms thickly on a new BCYE plate and incubating one day (if from a previous plate passage) or two to three days (if from frozen stock) to obtain confluent growth.
- Prepare BCYE plates by dissolving 10 g yeast extract, 10 g N-(2-acetamido)-2-aminoethanesulfonic acid, 0.35 g K2HPO4, and 1 g monobasic potassium α-ketoglutarate in 950 ml deionized water. Adjust to pH 6.9 with Potassium hydroxide (≥2.5 ml of 11.9 molar solution).
- Add 15 g agar and 2 g activated charcoal; and bring to 1 L final volume. Add a magnetic stir bar and autoclave. Cool medium to 55 °C and add filter-sterilized solutions containing 0.4 g L-cysteine and 0.42 g Ammonium iron(III) citrate dissolved in deionized water. Use magnet stir plate to mix prior to pouring Petri plates.
- For testing of L. pneumophila growth in axenic growth medium, prepare ACES-yeast extract broth (AYE) by combining all chemical reagents used for BCYE except for charcoal and agar. Filter sterilize and use immediately, or freeze for later experiments.
- Resuspend organisms in the same tissue culture medium used for J774A.1 cells with 100 µg/ml thymidine supplementation as appropriate.
- Passage L. pneumophila (Lp02::flaA::lux8) bacteria on appropriate medium, i.e., buffered charcoal yeast extract (BCYE) medium. If using a thymidine auxotrophic strain, supplement medium with 100 µg/ml thymidine. Prepare patches of bacteria for experiments by spreading organisms thickly on a new BCYE plate and incubating one day (if from a previous plate passage) or two to three days (if from frozen stock) to obtain confluent growth.
- Macrophage Infection
- Add test compounds of interest (screening compounds, and positive and negative controls for bacterial growth inhibition and eukaryotic cell lysis). Preferably, dissolve stock solutions at ≥500x in DMSO (dimethyl sulfoxide) or an aqueous solution to allow sufficient dilution of vehicle.
- Although stock solutions can be stored frozen, avoid freeze-thaw cycles for labile compounds and antimicrobials.
- Dilute luminescent bacteria to target of 2.5 x 106 CFU (colony forming unit)/m and fluorescent reporter-labeled bacteria to 1.0 x 107 CFU/ml in tissue culture medium and add appropriate non-toxic, membrane-impermeant, nucleic acid-binding dye at 2.5x final assay concentration.
- Add 20 µl of mixture to each J774A.1 culture well, final assay volume 50 µl, final bacterial concentration 1 x 106 CFU/ml (lux operon reporter) or 4 x 10 106 CFU/ml (fluorescent protein reporter).
- Incubate at 37 °C for 1-3 days in 5% CO2 at 100% relative humidity to prevent evaporative edge effects.
- Add test compounds of interest (screening compounds, and positive and negative controls for bacterial growth inhibition and eukaryotic cell lysis). Preferably, dissolve stock solutions at ≥500x in DMSO (dimethyl sulfoxide) or an aqueous solution to allow sufficient dilution of vehicle.
- Assay Readout
- Read bacterial growth and eukaryotic cell toxicity on a microplate luminometer and fluorimeter as appropriate for reporters being used.
- To avoid temperature-associated edge effects, thermally equilibrate microplates prior to luminescence reading by placement in a single layer on a lab bench with lids ajar for approximately 20 min (or in a biological safety cabinet with lids off for approximately 10 min).
- Return microplates to incubator if real-time readout at later time points is desired.
- Read bacterial growth and eukaryotic cell toxicity on a microplate luminometer and fluorimeter as appropriate for reporters being used.
2. Data Analysis
- Normalize data to positive and negative controls to calculate percent or fold-inhibition for intracellular bacterial growth and percent cytotoxicity for eukaryotic cells.
- To assess assay robustness, calculate statistical separation between positive and negative controls for both bacterial growth inhibition and cytotoxicity using Z-factor (Z') analysis where Z' = 1 – 3(σp + σn)/|µp + µn|. In this equation, σp and σn are the standard deviations for the positive and negative control wells, respectively; µp and µn are the mean values for the positive and negative control wells, respectively.
- For high-throughput screening assays, calculate standard z-scores (or other alternative, quantitative statistical measures) to assess effects of test compounds of interest. Alternatively, calculate a simple fold-reduction or log-fold reduction as a potentially physiologically more relevant measure of effect magnitude for geometrically replicating bacteria.
3. Single and Multi-dimensional (i.e., Synergy) Dose-response Testing and Data Interpretation
- Prepare macrophages and bacteria as described in protocol section 1.
- Just prior to infection or axenic incubation, add antimicrobials of experimental interest in a serial, doubling dilution series, with the goal of spanning on the high end a concentration that completely eliminates growth (i.e., the minimal inhibitory concentration or MIC) and on the low end a concentration that shows no obvious activity.
- Use an automated liquid handling system to facilitate single dose-response and combinatorial synergy testing setup through direct, automated addition of antimicrobial dilutions according to manufacturer's protocol.
- Consider use of sub-doubling dilutions, for example,
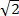 -fold dilution series, and assay replicates to increase accuracy and reproducibility of data.
-fold dilution series, and assay replicates to increase accuracy and reproducibility of data. - For macrophage infection, dilute luminescent bacteria to target of 2.5 x 106 CFU (colony forming unit)/ml in tissue culture medium. Add 20 µl of mixture to each J774A.1 culture well, final assay volume 50 µl, final bacterial concentration 1 x 106 CFU/ml; and incubate at 37 °C in 5% CO2 at 100% relative humidity.
- For axenic growth testing, dilute luminescent bacteria to final concentration of 1 x 106 CFU/ml in AYE medium, add 50 µl to each well of a 384-well plate and incubate at 37 °C in 5% CO2 at 100% relative humidity.
- Calculate fractional inhibitor concentration index for antimicrobial combinations that inhibit bacterial growth. For each drug in the combination (a, b, . . n) that results in complete or >99% inhibition of bacterial growth, calculate the individual fractional inhibitory concentration (FICa, b, . . n.) as follows: FICa = the concentration of compound "a" divided by the minimal inhibitor concentration of "a" by itself.. Using the same formula, calculate FICb, etc. Then calculate the combinatorial FIC index or
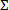 FIC = FICa + FICb + . . .FICn for each inhibitory combination.
FIC = FICa + FICb + . . .FICn for each inhibitory combination.
- Use the combinatorial index that deviates furthest from 1.0 to assess synergy. Consider a cutoff of ≤0.5 as a conservative indication of a synergistic effect and ≥4, an antagonist effect.12
- Plot isobolograms, isocontour isobolograms, and/or isosurface isobolograms6 as alternative graphical representations of combinatorial effects.
Microplate intracellular growth assay
Figure 1 diagrams the assay steps. The automated steps shown can be performed manually. However, throughput is greatly facilitated using liquid handling systems.
Figure 2 shows representative results from a 384-well microplate, dual-readout, real-time intracellular growth and eukaryotic cell cytotoxicity assay using a Legionella strain (Lp02) marked with either a lux operon (Figures 2A, 2B) or mNeptune2 fluorescent protein (Figure 2C, 2D) reporter. Positive and negative control compounds (DMSO, antibiotics, saponin) were added to plates through use of a pin transfer robot to simulate performance in a high-throughput screening setting. Significant Legionella growth (Figures 2A, 2C) can be easily distinguished in comparison to saponin lysis and antibiotic-treated controls at 24 to 72 hr for luminescent bacteria and 48 to 72 hr for mNeptune2-labeled bacteria, respectively. Legionella typically lyses host cells after replicating for 48 to 72 hr. This can be appreciated in Figures 1B and 1D, which show increased fluorescence of SYTOX Green (a representative, impermeant, nucleic acid-binding dye and marker of host cell death) over time. Cytotoxicity eventually reaches the maximal amount observed in the detergent (saponin), host cell lysis, positive control. mNeptune2-labeled organisms were added at a four-fold higher inoculum than lux operon-labeled bacteria, likely accounting for more rapid host cell killing and earlier associated rise in fluorescence signal in mNeptune2 experiments. As expected, effective antimicrobial treatment (levofloxacin or azithromycin) prevented both bacterial growth and host cell lysis. Conversely, cytotoxic compounds that limited intracellular growth through destruction of the host cell (e.g., saponin) could easily be identified by a combination of low luminescence or mNeptune2 fluorescence, and high cytotoxicity-associated signal. Furthermore, observation of combined decrease in both luminescence and mNeptune2 fluorescence (bacterial growth), and cytotoxicity provided reasonable confidence that a compound is a true intracellular growth inhibitor (e.g., azithromycin and levofloxacin positive controls) rather than a false positive resulting from spurious interference with bacterial reporter signal.
Table 1–3 shows representative Z' for three different bacterial reporters (lux operon, mNeptune2, tdTomato) and corresponding fluorescence readings relative to controls. A Z' >0.5 indicates a statistically robust assay appropriate for high-throughput settings; however, lower Z' may be suitable depending on experimental goals, i.e., the ability to reliably distinguish more or less subtle effects of test compounds or perturbations. Based on results from Figure 2, mNeptune2 was not as sensitive a reporter as the bacterial luciferase operon. Therefore, as expected, a robust difference between mNeptune2 signal in negative (DMSO) and positive (levofloxacin, azithromycin, saponin) growth inhibitor controls was not observed until day 2 or day 3 of incubation, in contrast to early, reasonably robust signal of lux operon reporter bacteria on day 1 after infection. In contrast, tdTomato, an alternative bacterial fluorescent reporter, showed suboptimal Z' throughout the infectious course. Suboptimal fluorescent characteristics resulted in part from encroachment of the fluorescence excitation and emission tail into the range used for optimal readout of tdTomato signal. This encroachment can be appreciated from the positive Z' found in the tdTomato saponin versus levofloxacin comparison (implying a significant difference in bacterial numbers under two conditions where bacteria should not replicate). This result can be explained by saponin lysis causing a strong fluorescence signal, whose tail is detected as tdTomato emission, and therefore leading to a spurious difference between the saponin and antibiotic controls. Therefore, while the bacterial lux and mNeptune2 reporters in combination with fluorescence appear usable for dual-readout, solution-based, real-time assays, the tdTomato and fluorescence combination, at least without further mathematical signal deconvolution, does not.
Figure 3 shows previously published examples of dose-response curves of known antimicrobials using lux operon, reporter-labeled organisms.6 Notably, Legionella does not grow in tissue culture medium. Therefore, replication of bacteria in macrophage infection assays solely represents intracellular growth. However, Legionella will grow axenically in a specialized, low sodium, liquid ACES (N-(2-acetamido)-2-aminoethanesulfonic acid) yeast extract medium.20 Here effects on intracellular and axenic growth were compared. Using these two growth systems, we observed that polar antimicrobials such as β-lactams (meropenem, ceftriaxone) and aminoglycosides (data not shown) are poor inhibitors of intracellular growth, in contrast to their potent effects on axenic growth. Presumably poor efficacy in macrophage infection assays is based on an inability to access the intracellular Legionella replicative niche. In contrast, eukaryotic cell-penetrant antimicrobials such as quinolones (levofloxacin) and azithromycin, demonstrated potent effects on both intracellular and axenic bacteria.
Additionally, use of the dual-readout assay allowed dose-response curves for both bacterial inhibition and eukaryotic cytotoxicity to be determined in the same experiment. Specifically, IC50 (concentration for 50% bacterial growth inhibition) and CC50 (concentration that induces 50% eukaryotic cell death) can be determined, and selectivity, CC50/IC50, calculated. All three measures are important criteria for compound progression in drug discovery efforts. Figure 4 shows an example of such dual, dose-response curves in response to the antibiotic doxycycline, data obtained from the same screening wells over time. In Figure 4A, on day 1 of infection, about a hundred-fold bacterial replication is evident in the zero antibiotic test wells compared with the highest levels of inhibition observed with antibiotic. There was minimal associated eukaryotic cell toxicity except at very high doxycycline concentrations (≥10 µg/ml). On day 2 (Figure 4B), bacterial lux signal was approximately 10-fold greater than day 1 with significant bacterial replication-associated cytotoxicity observed at low antimicrobial concentrations. As expected, bacterial replication-induced host cell death tracks closely with bacterial numbers (lux signal) throughout the antimicrobial dose-response curve. At higher antimicrobial concentrations cytotoxicity is reduced to baseline, until very high concentrations, where doxycycline again appears toxic as observed on day 1. The apparent shifting of the luminescence dose-response towards slightly higher antimicrobial concentrations compared to fluorescence-based cytotoxicity measurements is somewhat exaggerated when plotting luminescence and cytotoxicity on log and linear scales, respectively, as shown. IC50 for bacterial growth was approximately 100 ng/ml while CC50 was approximately 10 µg/ml, consistent with the CC50 described previously for doxycycline treatment of the leukocyte lineage, HL-60 cell line.21 Therefore, overall selectivity determined in this dual-readout experiment was approximately 100.
Figure 5 shows previously published isocontour isobolograms associated with two-dimensional synergy tests in which pairwise combinations of antimicrobials were tested for enhanced effect against intracellular L. pneumophila.6 Although standard isobolograms connect the lowest combinatorial concentrations of antibiotics that completely inhibit growth, we opted, based on available quantitative inhibitory data, to connect points with similar levels of inhibition using isocontours. The right most isocontour in these diagrams corresponds to the standard isobologram line. Concave isobolograms generally indicate synergy, while convex isobolograms generally indicate indifference or antagonism. Examples of synergy (panels A-C) and indifference are shown (D-F), in this case corresponding to FIC index values of <0.5 and ≥1, respectively.6
Of note, pairwise combinations of azithromycin, minocycline, and rifampicin demonstrated synergy. Therefore, these agents were tested together in three-way combination as shown in Figure 66. Here, a surface was drawn to connect the lowest combinatorial concentrations of the three antimicrobials that led to >99% inhibition of bacterial intracellular growth. The observed concave-shaped surface, by itself suggestive of three-dimensional synergy, corresponded to a low FIC index (0.325) indicative of substantial synergy effect.

Table 1: Z' for lux operon-reporter, Legionella infection- comparison of positive and negative controls. Z' correspond to data points shown in Figure 2A, 2B. This table has been modified from Chiaraviglio and Kirby 2014.4 Positive luminescence Z' >0.25 are highlighted with yellow characters on a black background.

Table 2: Z' for mNeptune2-reporter, Legionella infection — comparison of positive and negative controls. Z' correspond to data points shown in Figure 2C, 2D. Positive mNeptune2 fluorescence Z' >0.25 are highlighted with red characters.
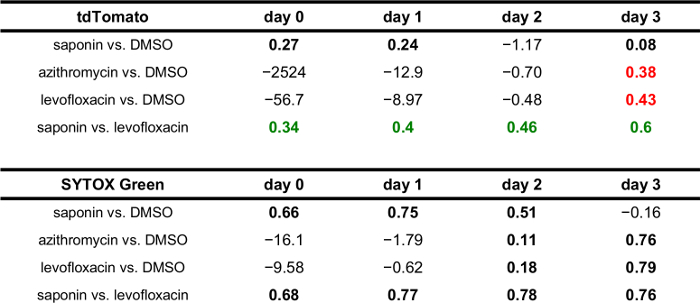
Table 3: Z' for tdTomato-reporter, Legionella infection — comparison of positive and negative controls. Positive tdTomato fluorescence Z' >0.25 are highlighted with red characters. False positive Z' >0.25 related to overlap of SYTOX Green and tdTomato emission are highlighted with green characters.

Figure 1: Pictorial representation of dual-reporter assay setup. (A) Replating of J774A.1 cells grown in suspension into 384-well dishes. (B) Addition of compounds of interest to microplates. (C) Simultaneous infection and addition of nucleic acid-binding dyes; incubation; and readout. Please click here to view a larger version of this figure.
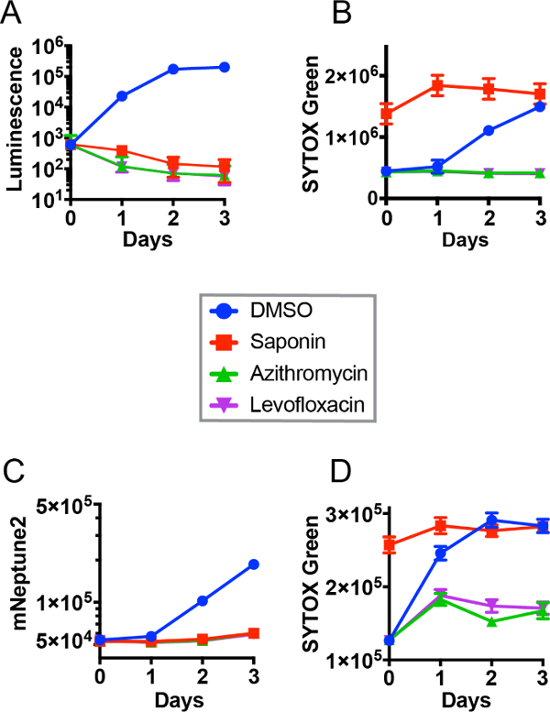
Figure 2: Dual, real-time readout of intracellular bacterial growth and eukaryotic cytotoxicity in high-throughput format. Corresponding luminescence (A) and fluorescent (B) signals during intracellular infection of J774A.1 cells with lux operon-expressing Legionella pneumophila. Corresponding fluorescent protein (C) and fluorescent (D) signals during intracellular infection of J774A.1 cells with mNeptune2-expressing Legionella pneumophila. Four treatments are shown: negative DMSO control ( ); positive cytotoxicity control, saponin (
); positive cytotoxicity control, saponin ( ); and positive bacterial growth inhibition controls, azithromycin (
); and positive bacterial growth inhibition controls, azithromycin ( ) and levofloxacin (
) and levofloxacin (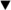 ). Data points represent mean and standard deviation of 96 replicates performed in duplicate, 384-well plates. Note, standard deviation error bars for some data points were minimal and therefore hidden within data point symbols. Panels A and B have been modified from Chiaraviglio and Kirby 2014.4 Please click here to view a larger version of this figure.
). Data points represent mean and standard deviation of 96 replicates performed in duplicate, 384-well plates. Note, standard deviation error bars for some data points were minimal and therefore hidden within data point symbols. Panels A and B have been modified from Chiaraviglio and Kirby 2014.4 Please click here to view a larger version of this figure.
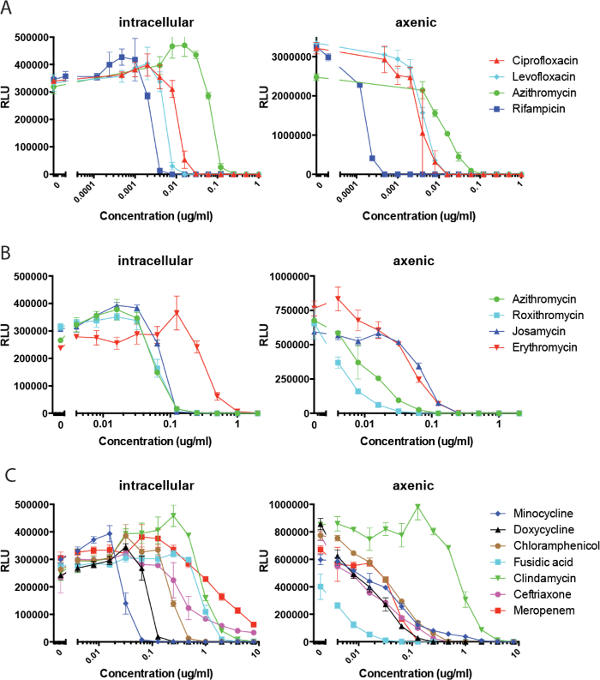
Figure 3: Representative examples of one-dimensional, dose-response assays. J774A.1 cells were infected with lux operon-expressing, L. pneumophila and treated with a two-fold dilution series of indicated antimicrobials (labeled as 'intracellular'). In parallel, lux operon-expressing, Legionella were diluted to the same final concentration in aces yeast extract medium (AYE) and treated with an identical, two-fold dilution series (labeled as 'axenic'). Luminescence was read two days post bacterial inoculation and plotted versus antimicrobial concentration. Panel A includes antimicrobials commonly used to treat Legionella infection. Panel B compares activity of different macrolide antibiotics. Panel C includes several different classes of antimicrobials with varying and sometimes contrasting intracellular and axenic potency. Data points represent the averages and standard deviations of three separate test wells per condition. This figure has been modified from Chiaraviglio and Kirby 2015.6 Please click here to view a larger version of this figure.
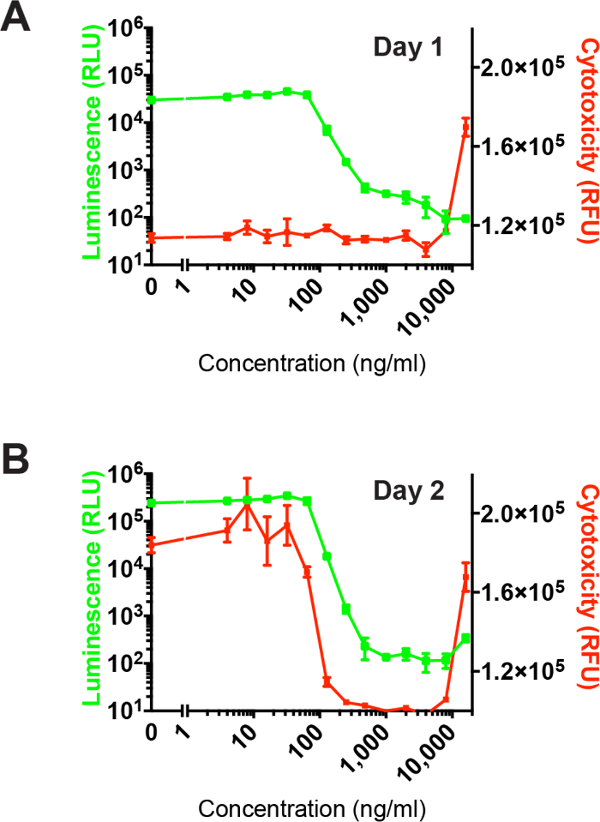
Figure 4: Example of a dual-readout, dose-response experiment for determining IC50, CC50 and selectivity. Effects of doxycycline on intracellular growth and J774A.1 cell cytotoxicity were determined on days 1 and 2 post infection. Data points represent the averages and standard deviations of three separate test wells per condition. Please click here to view a larger version of this figure.
Figure 5: Example of two-dimensional synergy assays. Combinatorial serial dilutions of pairs of antimicrobials were tested for synergy against intracellular growth of Legionella in J774A.1 macrophages. Isocontours lines connect points of similar percent inhibition. The rightmost isobologram connects combinations with complete intracellular growth inhibition (>99% luminescence reduction compared to untreated controls) and corresponds to the standard isobologram plot. Shading indicates percent inhibition as delineated by the color-coded key in each graph. Isocontours from left to right correspond to 100, 90, 80, 70, 60, 50, 40, 30, 20, 10, 5, and 1% growth relative to untreated controls. This figure has been modified from Chiaraviglio and Kirby 2015.6 Please click here to view a larger version of this figure.
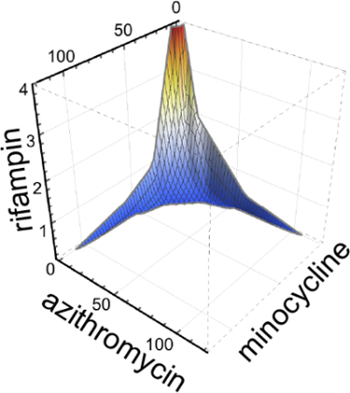
Figure 6: Example of a three-dimensional synergy assay. The lowest combinatorial concentrations of azithromycin, minocycline and rifampicin that resulted in >99% growth inhibition were connected to form an isobologram surface plot. This figure has been modified from Chiaraviglio and Kirby 2015.6 Please click here to view a larger version of this figure.
