In our experience, TIG-1 cells can be made almost completely tetraploid by simple continuous treatment with 0.1 µg/mL DC for 4 days (Figure 2A). In contrast, other fibroblast strains, such as BJ or IMR-90, and TIG-hT cells, became a mixture of diploid and tetraploid populations following the same treatment, and isolation of mitotic cells by the shake-off method is necessary during the course of DC treatment (usually 16 – 18 h after the start of treatment) (Figures 2B, C). After an additional treatment with DC for 3 days, cells underwent growth arrest and exhibited large, flattened morphology for several days. Within one week in general, small proliferating cells appeared among the large flattened cells (Figure 3). Cells became almost completely tetraploid in 2 – 3 weeks after DC treatment. The modal chromosome number in established tetraploid cells was 92 in most cases, except that one of the tetraploid cell lines established from TIG-hT cells had 91 chromosomes in the majority of cells (Figure 4). Established tetraploid cells grew at almost the same rate as diploid cells (Figure 5). Tetraploid cell lines established from non-immortalized cells showed an approximately equal replicative lifespan as the original diploid cells, while those from immortalized cells seemed to be also immortal (Figure 5). Tetraploid cells established from non-immortalized TIG-1 cells showed a normal karyotype except for having a tetraploid chromosome number (Figure 6 top panel), whereas those from immortalized TIG-1 (TIG-hT) cells showed rather complicated clonal aberrations (Figure 6 middle and bottom panels). The frequency of clonal aberrations in the latter cells (TIG-hT-4n) increased from 2.5 aberrations per cell at 255 population doubling levels (PDLs) to 5.2 aberrations per cell at 450 PDL with repeated subculture.
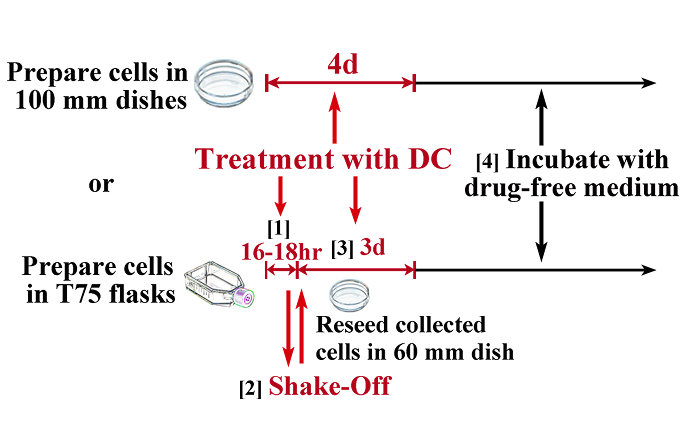
Figure 1: Schematic Illustration of the Protocol for Induction of Tetraploidy. To obtain tetraploid cells with minimum contamination of diploid cells, a combination of mitotic shake-off and DC treatment is necessary for most cell strains (lower illustration), while some cell strains (e.g. TIG-1) can become almost completely tetraploid by continuous treatment with demecolcine (upper illustration). 1) Treat cells in T75 flasks (at least 5 x 106) with 0.1 µg/mL DC for 16 – 18 h (this time should be determined by preliminary experiments) to arrest cells in mitosis. 2) Collect mitotic cells by the shake-off method and reseed into 60 mm culture dishes. 3) Treat cells with 0.1 µg/mL DC for an additional 3 days. 4) Incubate cells in drug-free medium (usually cells resume proliferation within 1 week). Scale bars represent 100 µm. Please click here to view a larger version of this figure.
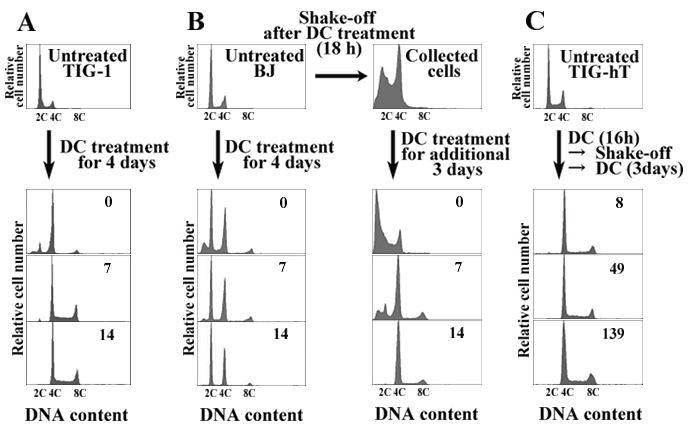
Figure 2: DNA Histograms of Fibroblasts Before and After Treatments for Induction of Tetraploidy. (A) TIG-1 cells. This cell strain becomes tetraploid by simple continuous treatment with 0.1 µg/mL DC for 4 days. (B) BJ cells. Because this cell strain becomes a mixture of diploid and tetraploid populations following simple treatment with DC (left panels), isolation of mitotic cells by shaking off is necessary during DC treatment to establish tetraploid cells (right panels). (C) TIG-hT (telomerase-immortalized TIG-1) cells. These cells also need to be prepared by a combination of DC treatment and shake-off for establishment of tetraploid cells. Numerals in the histograms represent the time (days) after drug removal. The abscissa represents DNA content (C, complement). Please click here to view a larger version of this figure.
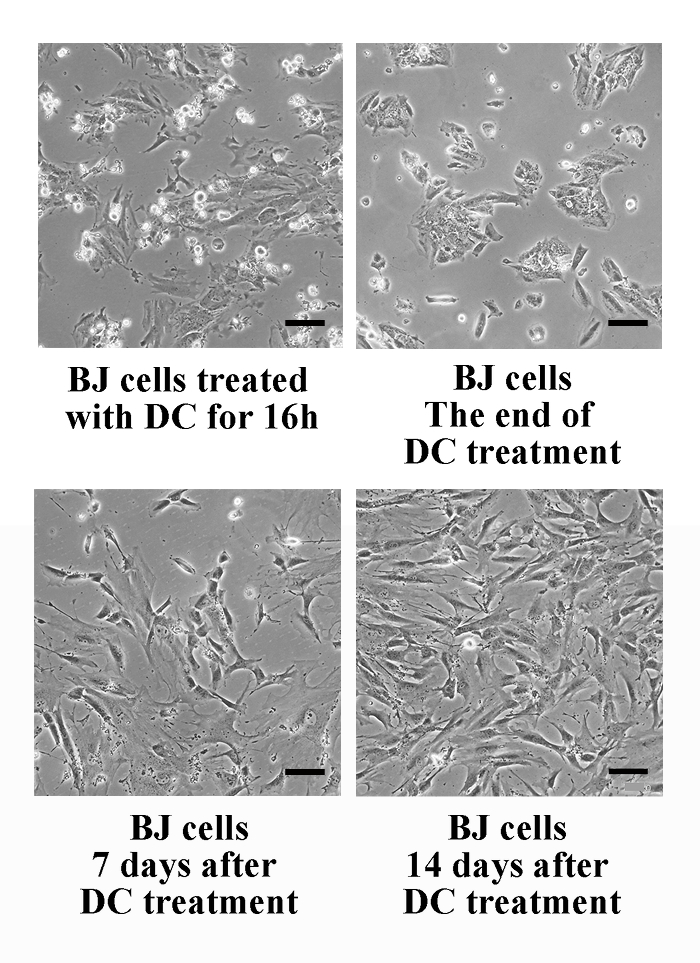
Figure 3: Representative Photomicrographs of BJ Cells Treated with DC. (A) BJ cells treated with 0.1 μg/mL DC for 16 h. Many cells arrested in mitosis can be seen. (B) The end of DC treatment. Almost all cells are growth-arrested. (C) 7 days after DC treatment. Small proliferating cells among large flattened cells can be seen. (D) 14 days after DC treatment. Cells are actively growing. Please click here to view a larger version of this figure.
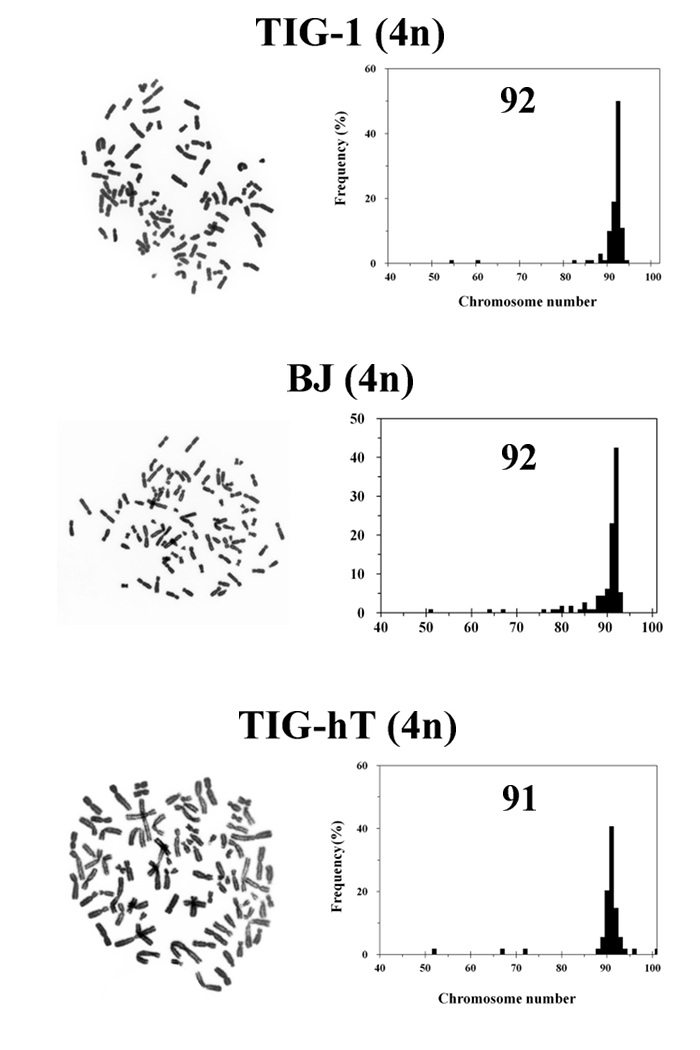
Figure 4: Representative Photomicrographs of Chromosomes and Histograms of Chromosome Number for Tetraploid Cells Established From Each Cell Strain. Top, middle and bottom panels represent tetraploid cells established from TIG-1 (2 weeks after DC treatment), BJ (2 weeks after DC treatment) and TIG-hT (7 weeks after DC treatment) cells respectively. Chromosomes were scored in at least 50 metaphases. Numerals in histograms are modal chromosome numbers. Please click here to view a larger version of this figure.
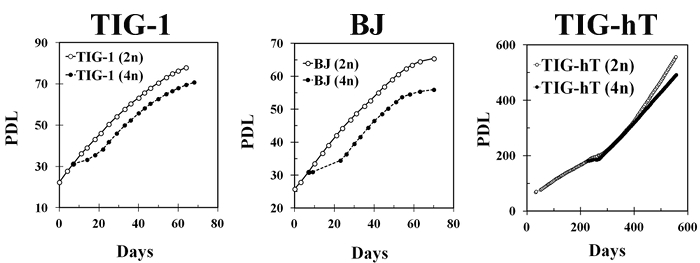
Figure 5: Representative Growth Profiles of Human fibroblasts (Diploid Cells) and Tetraploid Cells Established from Each Cell Strain. Left, center and right panels show growth profiles of original TIG-1, BJ and TIG-hT cells (open markers) and those of tetraploid cells established from each cell strain (closed markers). Cells were passaged twice a week and PDLs were calculated from cell numbers. Please click here to view a larger version of this figure.

Figure 6: Representative Karyograms by G-banding and mFISH. Top panel shows a karyogram by G-banding of tetraploid cells established from TIG-1 (2 weeks after DC treatment). Middle and bottom panels show karyograms by mFISH for tetraploid cells established from TIG-hT cells (15 and 41 weeks after DC treatment). Karyotypes were based on 10 cells (TIG-1) or 20 cells (TIG-hT). Please click here to view a larger version of this figure.
