Use of a Monocyte Monolayer Assay to Evaluate Fcγ Receptor-mediated Phagocytosis
Summary
The monocyte monolayer assay (MMA) is an in vitro assay that utilizes isolated primary monocytes obtained from mammalian peripheral whole blood to evaluate Fcγ receptor (FcγR)-mediated phagocytosis.
Abstract
Although originally developed for predicting transfusion outcomes of serologically incompatible blood, the monocyte monolayer assay (MMA) is a highly versatile in vitro assay that can be modified to examine different aspects of antibody and Fcγ receptor (FcγR)-mediated phagocytosis in both research and clinical settings. The assay utilizes adherent monocytes from peripheral blood mononuclear cells isolated from mammalian whole blood. MMA has been described for use in both human and murine investigations. These monocytes express FcγRs (e.g., FcγRI, FcγRIIA, FcγRIIB, and FcγRIIIA) that are involved in immune responses. The MMA exploits the mechanism of FcγR-mediated interactions, phagocytosis in particular, where antibody-sensitized red blood cells (RBCs) adhere to and/or activate FcγRs and are subsequently phagocytosed by the monocytes. In vivo, primarily tissue macrophages found in the spleen and liver carry out FcγR-mediated phagocytosis of antibody-opsonized RBCs, causing extravascular hemolysis. By evaluating the level of phagocytosis using the MMA, different aspects of the in vivo FcγR-mediated process can be investigated. Some applications of the MMA include predicting the clinical relevance of allo- or autoantibodies in a transfusion setting, assessing candidate drugs that promote or inhibit phagocytosis, and combining the assay with fluorescent microscopy or traditional Western immunoblotting to investigate the downstream signaling effects of FcγR-engaging drugs or antibodies. Some limitations include the laboriousness of this technique, which takes a full day from start to finish, and the requirement of research ethics approval in order to work with mammalian blood. However, with diligence and adequate training, the MMA results can be obtained within a 24-h turnover time.
Introduction
The monocyte monolayer assay (MMA) is an in vitro assay originally developed to better predict blood transfusion outcomes in patients with auto- or alloantibodies to red blood cells (RBCs)1-5. By assessing the effect of anti-RBC antibodies in mediating Fcγ receptor (FcγR)-mediated phagocytosis using this in vitro assay, it is possible to predict the clinical outcome in vivo. Indeed, the MMA has been used successfully to avoid immune destruction of antibody-bound RBCs, despite the transfusion of serologically incompatible blood5. The typical pre-transfusion procedure for compatibility testing, also termed crossmatching, involves serological methods that include typing the patient’s blood for ABO and Rh antigens and screening for the presence of anti-RBC antibodies in the patient6. Blood matched for ABO/Rh is selected, and if antibodies are present, an attempt to identify them is made so that blood for transfusion can be further selected to avoid these antigens. An ideal crossmatch result occurs when all donor blood is serologically compatible with the patient’s blood, which reduces the risk of post-transfusion hemolysis7. However, this system falls short for the small group of patients who have become alloimmunized upon repeated transfusion or pregnancy. These patients produce alloantibodies against specific RBC antigens. Some produce antibodies to antigens of very high frequency in the general population, and thus become progressively more difficult to crossmatch8,9. Adding to the complexity, not all alloantibodies are clinically significant; in other words, the binding of an alloantibody to RBCs detected by a serology test does not necessarily result in hemolysis when antigen-positive, incompatible blood is transfused. The MMA was originally developed to assess the potential clinical significance of serologically incompatible blood in a transfusion setting1-5.
Since extravascular hemolysis of antibody-bound RBCs is known to be mediated by the mononuclear phagocyte system, primary monocytes/macrophages are utilized in the development of diagnostic assays. The first assay to study the interaction of monocytes, RBCs, and antibodies was published in 1975, but the sub-optimal conditions used led only to rosette formation (the binding of RBCs to the periphery of the monocyte), and no phagocytosis was observed10. Significant modifications to the assay were made by several groups, leading to an assay for which the level of phagocytosis of alloantibody-bound RBCs could be correlated to the clinical outcome of hemolysis1-5. Recently, the optimal storage conditions of clinical samples and further optimization of assay conditions were examined to enhance the utility of a clinical MMA crossmatch using autologous patient samples11.
Three other diagnostic techniques have been employed in addition to the MMA in predicting transfusion outcomes: the 51Cr release test, the rosette test, and the chemiluminescence test (CLT). In the 51Cr release test, the patient is injected with 51Cr-labeled donor RBCs, and the half-life of the labeled RBCs is monitored and is predictive of post-transfusion survival or clearance12,13. As this method uses radioactive materials, it is rarely performed anymore. The rosette test involves mixing and incubating monocytes with RBCs and quantifying the level of rosette formation (with no phagocytosis)14. The clinical significance of antibodies in vivo involves active phagocytosis by macrophages found in the spleen and/or the liver; thus, this method does not provide a relevant readout of phagocytosis. The CLT uses luminol to monitor the oxidative burst during monocyte phagocytosis of RBC, since luminol fluoresces blue when oxidized in the phagosome15. This method is good, but contamination by neutrophils can confound the readout. Parallel comparisons have been made to evaluate the sensitivity, practicality, and reproducibility of the four available methods, and both the CLT and MMA were ranked superior16. However, the CLT has been mainly utilized in assessing hemolytic disease of the fetus and newborn (HDFN), and the assay’s optimal pH of 8.0 might compromise the level of phagocytosis11.
In addition to its diagnostic and clinical utility, the MMA has been modified for other research purposes. Indeed, the MMA can not only serve as a functional assay to address discrepancies between serology and biology, it has also been used to retrospectively investigate the cause of hemolysis after intravenous immunoglobulin (IVIG) therapy17. It has also been used to examine the structure-function of chemical inhibitors of FcγR-mediated phagocytosis18-20 and to study the downstream signaling of FcγR-mediated phagocytosis21. In our laboratory, in addition to using a human MMA, we are developing a murine MMA using primary mouse peripheral blood mononuclear cells (PBMCs) and autologous RBCs. The rationale is to screen antibodies that can induce FcγR-mediated phagocytosis as an intermediate to developing an in vivo autoimmune hemolytic anemia (AIHA) mouse model (unpublished data). The various modifications focus on different aspects of the IgG antibody and FcγR interaction that induce phagocytosis.
Protocol
Obtain approval for the use of human samples by the research ethics board/institutional review board, and obtain signed consent from all human donors. The MMA can also be performed using mouse PBMCs in a similar manner as the human assay, following animal use ethics approval. The MMA utilizes aseptic tissue culture techniques.
NOTE: See Figure 1.
NOTE: Although we recommend use of a level 2 biocontainment cabinet during the assay to maintain aseptic technique, as the method is only a “short-term” culture method, there isn’t really enough time for bacteria to contaminate the assay. Therefore, if a level 2 biocontainment cabinet doesn’t exist, rather than buy one just for this assay, the assay could be performed outside of a biocontainment cabinet, on an open bench. The use of a 37 °C incubator with 5% CO2, however, is not an option and must be used for optimal results.
1. Peripheral Blood Mononuclear Cell Isolation
- Obtain human whole blood from a healthy donor or a patient via venipuncture using vacutainers containing acid-citrate-dextrose (ACD) anticoagulant (yellow-top tubes).
NOTE: Whole blood can be stored in ACD at room temperature (18-22 °C) for up to 36 h before proceeding to the next step11. Normally, 1-2 10-mL vacutainer tubes of whole blood are sufficient for the assay. - Dilute the whole blood 1:1 v/v in warm complete RPMI medium (RPMI-1640 supplemented with 10% fetal bovine serum, 20 mM HEPES, and 0.01 mg/mL gentamicin).
- Isolate peripheral blood mononuclear cells (PBMCs) from the diluted whole blood using density gradient centrifugation, as recommended by the manufacturer (See List of Materials). Layer the diluted blood very slowly over the density gradient (warmed to room temperature, 18-22 °C).
NOTE: Minimize the amount of mixing at the interface for the optimal separation of blood by carefully layering the blood mixture in a dropwise fashion or by using a pipette.- Allow the blood mixture to slowly layer over the top of the density gradient by placing the pipet tip close to the density gradient and by enabling the blood mixture to run down the side of the tube very slowly.
- Depending on the scale of experiment, layer 10 mL of blood mixture on top of 3 mL of density gradient (in a 15-mL tube) or layer 35 mL of blood mixture on top of 15 mL of density gradient (in a 50-mL tube). Typically, 10 mL of whole blood yields 10 million PBMCs, with some donor-to-donor variation.
NOTE: It is very important that there is no mixing of the blood mixture with the density gradient. The blood mixture should layer over the density gradient and slowly rise until all the blood is on top of the density gradient. - Centrifuge the layered mixture at 700 x g for 30 min without brakes. The centrifuged mixture should separate into 5 layers (from top to bottom): plasma, buffy coat (containing PBMCs), density gradient material, granulocytes, and RBCs.
- Remove and discard the majority of the plasma and carefully retrieve the buffy coat (PBMC) content into a new 15-mL tube using a Pasteur pipette and a suction bulb.
NOTE: The removal of the buffy coat layer is effectively done by applying suction on the Pasteur pipette while performing a circular motion around the outside of the layer, with the tip of the pipette against the tube.
- Wash the isolated PBMCs three times in pH 7.3 phosphate-buffered saline (PBS) by centrifuging at 350 x g for 10 min (with full brakes) in between washes. Reconstitute the PBMC pellet in complete RPMI medium. Depending on the size of the pellet, 3-7 mL of medium is sufficient.
- Count the PBMC using trypan blue and a hemocytometer. Only count those cells that are not stained by the trypan blue. Reconstitute the PBMCs to 1,750,000 cells/mL in complete RPMI medium.
- Seed 400 µL (700,000 cells) into each well of an 8-chamber slide. Incubate the slide in a 37 °C, fully humidified tissue culture incubator (supplemented with 5% CO2) for 1 h to allow the monocyte/macrophages to adhere.
2. Pre-treatment of Adhered Monocytes
NOTE: This step is only necessary if looking for ways to inhibit or enhance phagocytosis.
- Pre-treat adhered monocytes with any drug(s) or compound(s) of interest. Reconstitute the drug(s) or other test material to the desired concentration using complete RPMI medium.
NOTE: For example, 200 µg/mL of IVIG is typically used to yield a 95-100% inhibition of phagocytosis when using human monocytes. - Aspirate and discard the supernatant containing any non-adherent cells from the 8-chamber slide after the 1-h incubation (after step 1.6). Replace it with 400 µL of a drug or another treatment and incubate for 1 h at 37 °C.
- When aspirating and replacing solutions in the 8-chamber slide, make sure to control the flow of the fluids so that weakly adhered cells do not lift off. Also, work with only 2-3 empty wells at a time and avoid drying the wells.
NOTE: Typically, each treatment is performed in technical triplicates.
- When aspirating and replacing solutions in the 8-chamber slide, make sure to control the flow of the fluids so that weakly adhered cells do not lift off. Also, work with only 2-3 empty wells at a time and avoid drying the wells.
3. Opsonization of R2R2 Red Blood Cells
NOTE: The R2R2 used here are obtained from the Blood Collection Center (Canadian Blood Services), but they are also commercially available. Opsonized R2R2 RBCs are used as a positive control for FcγR-mediated phagocytosis. Naïve R2R2 should be stored in Alsever's solution (to prolong shelf life) at 4 °C for up to 1 month. Alsever's solution is made in-house and composed of 0.8% w/v trisodium citrate (dihydrate), 1.9% w/v dextrose, 0.42% w/v sodium chloride, and 0.05% w/v citric acid (monohydrate). If R2R2 cells are not available, other Rh-phenotyped cells, such as R1R2, R1R1, R1r, or R2r, can be used.
- Wash R2R2 (cDE/cDE) red blood cells in PBS a total of three times using centrifugation at 350 x g for 5 min each.
NOTE: The amount of RBCs needed depends on the experimental size and can be back-calculated. Always wash an excess amount of RBCs, since RBCs are lost during each wash due to lysis or during the removal of supernatant. For example, in an experiment with 5 treatments and 2 controls, all conducted in technical triplicates, there is a total of 21 wells. 21 wells with 400 µL/well of 1.25% RBC mixture indicates that 105 µL of packed, opsonized RBC are needed. 200 µL of RBC should be initially washed and 150 µL should be opsonized to ensure that there is an adequate amount of RBCs for the phagocytosis step. - Opsonize the washed R2R2 pellet with 1:1 v/v polyclonal anti-D antibodies from human serum and incubate for 1 h at 37 °C, with intermittent mixing.
NOTE: If polyclonal anti-D antibodies are not available, it is possible to use monoclonal anti-D antibodies, which are commercially available, or anti-D that is normally used for Rh immune prophylaxis, which can be obtained from blood banks or transfusion services. Optimization testing should be conducted at the beginning, when setting up the assay, and whenever switching lots of un-titered polyclonal antibodies or switching to a monoclonal antibody. This is to identify the optimal concentration or volume of anti-D required for an antiglobulin test (IAT) result of between 3+ and 4+ and a phagocytosis result of between 70-90 phagocytosed RBCs per 100 monocytes counted. - Wash the opsonized R2R2 a total of three times in PBS using centrifugation at 350 x g for 5 min each.
NOTE: Successful opsonization of R2R2 can be confirmed by performing an indirect IAT. Briefly, secondary polyclonal anti-human antibodies are added to bind primary opsonizing antibodies on RBC surfaces, and the amplified signal can be observed in the form of hemagglutination. A detailed manufacturer protocol can be found in the supplementary materials file. - Reconstitute the washed R2R2 pellet to 1.25% v/v using complete RPMI medium.
NOTE: Excess opsonized R2R2 can be stored in Alsever's solution at 4 °C for up to a week.
4. Fc Receptor-mediated Phagocytosis
- Aspirate the drug or medium supernatant from the 8-chamber slide and add 400 µL of the 1.25% v/v R2R2 mixture. Incubate at 37 °C for 2 h.
- After 2 h of incubation, remove the chambers using the manufacturer's adaptors. Dab off excess R2R2 on a paper towel. Make sure that the slide does not dry out.
- Fill a 100-mL beaker with PBS. Submerge and wash the slide by slowly moving the slide back and forth (around 30-40 strokes) to remove the majority of the un-phagocytosed R2R2.
- Remove the slide from the PBS. Dab off excess PBS using a paper towel or tissue and air-dry the slide.
- Fix the air-dried slide in 100% methanol for 45 s. Then, air-dry the fixed slide.
NOTE: Slides can be fixed using another method that is more compatible for downstream staining, such as the Grünwald-Giemsa stain21 or the Wright-Giemsa stain22. - Mount the slide using an in-house-made elvanol mounting medium (or another commercially available mounting medium) and add coverslips.
NOTE: Elvanol mounting medium is composed of 15% w/v polyvinyl alcohol resin and 30% v/v glycerin in PBS. The mixture is heated until all the resin has dissolved and the glycerin is homogenously mixed. This can be replaced with other commercially available mounting medium. - Allow the mount to dry overnight before quantification.
5. Quantification of Phagocytosis
- Using a phase-contrast microscope and a 40X objective lens, manually quantify the amount of phagocytic events by counting at least 200 monocytes and the number of phagocytosed R2R2 within these monocytes. Have one counter in each hand to simultaneously quantify the number of monocytes and the number of phagocytosed R2R2.
- Obtain the average phagocytic index by dividing the number of phagocytosed R2R2 by the number of monocytes and multiplying by 100. Express the data as the mean (average phagocytic index) ± the standard error of the mean (SEM).
Representative Results
By following the crucial steps in Figure 1 and the procedure outlined above, the MMA can be reproducibly performed. IVIG was used as an example for the inhibition of phagocytosis in Figure 2. IVIG is known to bind and block the Fc receptors, thus inhibiting the downstream outcome of phagocytosis. By titrating the amount of IVIG used, a dose-dependent inhibition is observed, in which concentrations above 200 µg/mL resulted in close to 100% inhibition and concentrations below 0.5 µg/mL did not inhibit at all (Figure 2A). When the phagocytic indices are transformed and normalized to the R2R2 positive control as 0% inhibition, an inhibition curve with an IC50 of 3 µg/mL can be determined (Figure 2B).
In addition to performing the assay consistently, quantification of the assay can sometimes be difficult. Figure 3 is a collection of phase-contrast microscopy images of various MMA slides. With microscopy experience, one can distinguish a monocyte from a contaminating RBC (Figure 3D), or a vacuole from a phagocytosed RBC (Figure 3B). Dense clusters of cells and/or debris should be avoided during quantification (Figures 3E and F). Also, one should avoid over-opsonizing the R2R2 RBC, which would lead to heightened phagocytosis that overcrowds the monocyte interior and interferes with accurate quantification (Figure 3C).
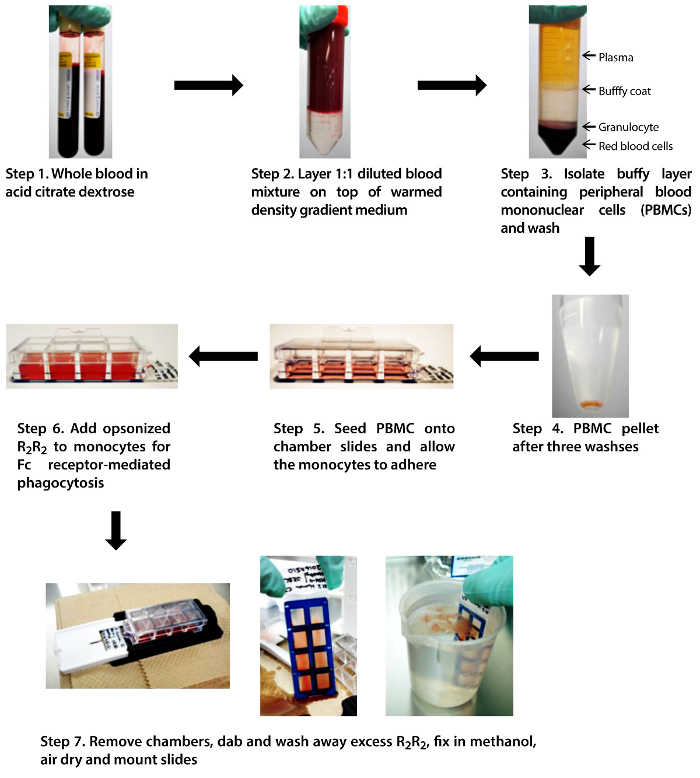
Figure 1. Schematic diagram of the MMA. A step-by-step depiction of the crucial steps in the MMA: isolating PBMC from whole blood, feeding the adhered monocytes with opsonized R2R2, and washing the chamber slides. Please click here to view a larger version of this figure.
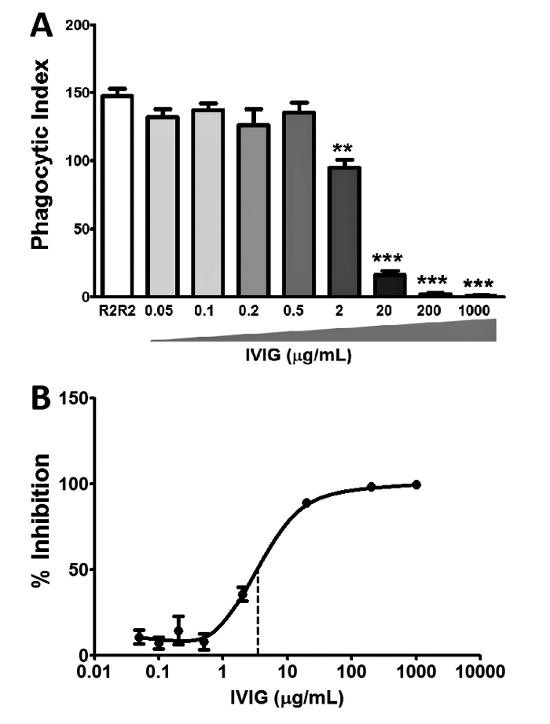
Figure 2. Intravenous IgG (IVIG) inhibits phagocytosis in vitro using the MMA. IVIG is known to inhibit phagocytosis and is used as an inhibition control in the human MMA. (A) Titration of IVIG resulted in a dose-dependent inhibition of phagocytosis when compared to the untreated R2R2 control. The results show the mean ± the standard error of the mean (SEM) of n = 3 experiments. The statistical analysis was conducted using Student t-test: ** P≤0.01 and *** P≤0.001. (B) Inhibition curve of IVIG with an IC50 of 3 µg/mL (dotted line). The results show data normalized to untreated R2R2 (0% inhibition); mean ± SEM of n = 3 experiments. Please click here to view a larger version of this figure.
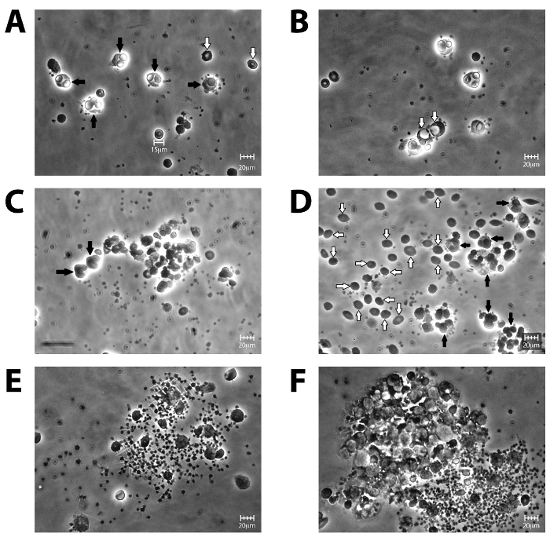
Figure 3. Phase-contrast microscopy images of sample slides under 40X magnification. (A) The perfect slide in which the monocytes phagocytosed 1-2 R2R2 on average (black arrows with a distinctive halo glow), with very few contaminating, un-phagocytosed R2R2 in the background (white arrows). (B) As shown in this slide, monocytes sometimes have enlarged vacuoles (white arrows), which can be mistaken for phagocytosed R2R2. (C) When R2R2 have been over-opsonized, more than 4-5 phagocytosed R2R2 per monocyte (black arrows) render accurate counting difficult, since the R2R2 are crowded within the monocyte and distinct cell boundaries cannot be distinguished. (D) Inadequate washing of the slide leads to abundant R2R2 contamination (white arrows), which could be mistaken for adhered RBCs. (E) Sometimes, monocytes and contaminating RBCs may form clusters. Clusters also indicate bacterial contamination, which should be avoided. (F) Monocytes may form larger aggregates, which should be avoided during quantification. Using random selection of the fields to view, panel A is what is usually observed if the MMA has been performed properly. Scale bar is 20 µm. Please click here to view a larger version of this figure.
Discussion
The MMA is a laborious technique that requires expertise in both tissue culture and microscopy. There are several critical steps to ensure success: 1) generation of the monocyte monolayer; 2) opsonization of the RBCs, and 3) manual quantification. The monocyte monolayer does not adhere very strongly to the chamber slide, so physiological pH must be maintained throughout the assay11 and an adequate number of PBMCs should be seeded. Vigorous pipetting, which might disturb the adhered cells, should be avoided. One approach is to always remove and add solutions from the same corner of the chamber and to ensure that the motion is slow and steady. Similarly, during the last washing step to remove the excess RBCs, the movement should be slow and steady. This ensures minimal disturbance to the monolayer while still removing the majority of the un-phagocytosed RBCs. Inadequate washing will lead to a high background of contaminating RBCs, which makes manual quantification difficult. Secondly, the R2R2 RBCs must be sufficiently opsonized to obtain an average phagocytic index of 80-120 for the phagocytosis control. This desired phagocytic range strikes a balance between the ease of counting (e.g., monocytes with more than 5 phagocytosed RBCs are difficult to accurately quantify) and maintaining an adequate amount of phagocytosis for statistical analysis. The degree of opsonization can be confirmed by an IAT, and a reading between 3+ to 4+ is needed. The R2R2 RBCs should be discarded when there is excess lysis during washing, when the supernatant turns dark red, or when a significant decrease in phagocytosis is observed in experiments due to the aging of the cells in storage. Lastly, manual quantification using the microscope can be tricky, especially when comparing counts between lab personnel and between experiments. By examining the same field on each well, or simply by counting more cells, a more consistent count can be obtained. Side-by-side training with an experienced technician and the use of a designated set of training slides is recommended.
A major critique of the MMA is the subjectivity of the manual quantification step. However, with adequate training, consistency can be obtained across different counters. Another limitation is the intrinsic donor-to-donor differences in monocyte phagocytic abilities and in R2R2 surface antigen expression levels, which is a source of data variation when dealing with human samples.
Other alternative techniques are available for examining FcγR-mediated phagocytosis. The majority of commercial kits utilize fluorescent output to monitor phagocytosis (e.g., bioparticles, pH-sensitive fluorescence protein, or IgG-labeled fluorescent latex beads). The use of fluorescent output does offer more objective quantification, but one also needs to consider the availability, cost, and training associated with the use of a fluorescent microscope or a flow cytometer, as well as the ensuing dependence upon commercially available kits.
Finally, this assay can be modified depending upon the research question. For example, when testing drug inhibition of phagocytosis, monocytes can either be pre-treated or co-incubated with both the drugs and opsonized RBCs (i.e., a competition assay). The downstream signaling of antibodies of different subtypes, chimeric antibodies, or recombinant constructs can also be tested. With recent breakthroughs in the development of a universal antigen-null blood24, the MMA could be utilized in initial screens of these antigen-null RBCs with various antibodies to assess whether there is indeed a lowered efficacy in triggering phagocytosis.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
The authors thank the Canadian Blood Services for a Graduate Fellowship Program Award to T.N.T. This research received financial support from the Canadian Blood Services’ Centre for Innovation, funded by the federal government (Health Canada) and the provincial and territorial ministries of health. The views herein do not reflect the views of the federal, provincial, or territorial governments in Canada.
Materials
| Acid citrate dextrose (ACD) vacutainers | BD | REF364606 | |
| RPMI 1640 | Sigma | R8758 | |
| HEPES | Bioshop | HEP003.100 | |
| Fetal bovine serum | Multicell | 080150 | |
| Gentamicin | Gibco | 15710-64 | |
| Ficoll-Paque PLUS | GE | 17-1440-03 | https://www.gelifesciences.com/ |
| Phosphate buffered saline | Sigma | D8537 | |
| 8-chamber slides | Lab-Tek-ll | 154534 | |
| R2R2 (cDE/cDE) red blood cells | Canadian Blood Services | Commercially available (e.g. http://www.bio-rad.com/en-ca/product/reagent-red-blood-cells) | |
| Polyclonal anti-D from human serum | Gamma Biologics | DIN 02247724 | Can be substituted with commercially available monoclonal anti-D or with Rh immune globulin |
| 100% methanol | Caledon | 6700-1-42 | |
| Polyvinyl alcohol resin | Sigma | P8136 | Can be substituted with commercially available mount |
| UltraPure glycerine | Invitrogen | 15514-011 | |
| Cover slips | VWR | 48366 067 | |
| Novaclone anti-IgG | Immucorgamma | 5461023 | Optional for IAT (http://www.fda.gov/downloads/biologicsbloodvaccines/…/ucm081743.pdf) |
Riferimenti
- Hunt, J. S., Beck, M. L., Hardman, J. T., Tegtmeier, G. E., Bayer, W. L. Characterization of human erythrocyte alloantibodies by IgG subclass and monocyte interaction. Am J Clin Pathol. 74 (3), 259-264 (1980).
- Schanfield, M. S., Stevens, J. O., Bauman, D. The detection of clinically significant erythrocyte alloantibodies using a human mononuclear phagocyte assay. Transfusion. 21 (5), 571-576 (1981).
- Branch, D. R., Gallagher, M. T., Mison, A. P., Sysiokian, A. L., Petz, L. D. In vitro determination of red cell alloantibody significance using an assay of monocyte-macrophage interaction with sensitized erythrocytes. Br J Haematol. 56 (1), 19-29 (1984).
- Hunt, J. S., Beck, M. L., Wood, G. W. Monocyte-mediated erythrocyte destruction. A comparative study of current methods. Transfusion. 21 (6), 735-738 (1981).
- Noumsi, G. T., Billingsley, K. L., Moulds, J. M. Successful transfusion of antigen positive blood to alloimmunised patients using a monocyte monolayer assay. Transfus Med. 25 (2), 92-100 (2015).
- Moulds, J. M. Introduction to antibodies and complement. Transf Apher Sci. 40 (3), 185-188 (2009).
- Grandstaff Moulds, M. K. Antibody identification. Transf Apher Sci. 40 (3), 195-197 (2009).
- Hendrickson, J. E., Tormey, C. A., Shaz, B. H. Red blood cell alloimmunization mitigation strategies. Transfus Med Rev. 28 (3), 137-144 (2014).
- Hamilton, J. R. Common and frequently encountered antibodies. Transfus Apher Sci. 40 (3), 189-194 (2009).
- Kenna, M. A., Cooper, R. A., Schrieber, A. D. Effect of papain on the interaction between human monocytes, erythrocytes and IgG. Blood. 46 (2), 245-252 (1975).
- Tong, T. N., et al. Optimal conditions for the performance of a monocyte monolayer assay. Transfusion. , (2016).
- Gray, S. J., Sterling, K. The tagging of red cells and plasma proteins with radioactive chromium. J Clin Invest. 29 (12), 1604-1613 (1950).
- Mollison, P. L., Veall, N. The use of the isotope 51Cr as a label for red cells. Br J Haematol. 1 (1), 62-74 (1955).
- Sebring, E. S., Polesky, H. F. Detection of fetal hemorrhage in Rh immune globulin candidates. A rosetting technique using enzyme-treated Rh2Rh2 indicator erythrocytes. Transfusion. 22 (6), 468-471 (1982).
- Downing, I., Templeton, J. G., Mitchell, R., Fraser, R. H. A chemiluminescence assay for erythrophagocytosis. J Biolumin Chemilumin. 5 (4), 243-250 (1990).
- Fabron, A., et al. Application of noninvasive phagocytic cellular assays using autologous monocytes to assess red cell alloantibodies in sickle cell patients. Transfus Apher Sci. 31 (1), 29-35 (2004).
- Michelis, F. V., et al. Acute hemolysis after intravenous immunoglobulin amid host factors of ABO-mismatched bone marrow transplantation, inflammation, and activated mononuclear phagocytes. Transfusion. 54 (3), 681-690 (2014).
- Rampersad, G. C., et al. Chemical compounds that target thiol-disulfide groups on mononuclear phagocytes inhibit immune mediated phagocytosis of red blood cells. Transfusion. 45 (3), 384-393 (2005).
- Purohit, M. K., et al. Structure-activity relationships of pyrazole derivatives as potential therapeutics for immune thrombocytopenias. Bioorg Med Chem. 22 (9), 2739-2752 (2014).
- Neschadim, A., Kotra, L. P., Branch, D. R. Small molecule phagocytosis inhibitors for immune cytopenias. Autoimmun Rev. 15 (8), 843-847 (2016).
- Fitzer-Attas, C. J., et al. Fcgamma receptor-mediated phagocytosis in macrophages lacking the Src family tyrosine kinases Hck, Fgr, and Lyn. J. Exp Med. 191 (4), 669-682 (2000).
- Allhorn, M., et al. The IgG-specific endoglycosidase EndoS inhibits both cellular and complement-mediated autoimmune hemolysis. Blood. 115 (24), 5080-5088 (2010).
- Li, L., et al. Inhibition of phagocytic recognition of anti-D opsonized Rh D+ RBC by polymer-mediate immunocamouflage. Am J Hematol. 90 (12), 1165-1170 (2015).
- Kwan, D. H., et al. Toward efficient enzymes for the generation of universal blood through structure-guided directed evolution. J Am Chem Soc. 137 (17), 5695-5705 (2015).
