Interaction between molecules is the basis of nature. Hence, scientists in many fields of basic and applied research try to understand the fundamental principles of molecular interactions of different kinds. MicroScale Thermophoresis (MST) enables scientists to perform the fast, precise, cost-efficient, and quality-controlled characterization of molecular interactions in solution, with a free choice of buffers. There are already more than 1,000 publications using MST, from 2016 alone, describing different kinds of analyses, including library screenings, binding event validations, competition assays, and experiments with multiple binding partners1-8. In general, MST permits the study of the classical binding parameters, such as binding affinity (pM to mM), stoichiometry, and thermodynamics, of any kind of molecular interaction. A great advantage of MST is the ability to study binding events independent of the size of the interaction partners. Even challenging interactions between small nucleic acid aptamers (15-30 nt) and targets such as small molecules, drugs, antibiotics, or metabolites can be quantified.
Current state-of-the-art technologies to characterize aptamer-target interactions are either lab-intense and highly complex or fail to quantify aptamer-small molecule interactions9,10. Surface Plasmon Resonance (SPR)-based assays11,12 and truly label-free calorimetric approaches, such as Isothermal Titration Calorimetry (ITC)13-15, isocratic elution16, equilibrium filtration17,18, in-line probing19, gel-shift assays, stopped-flow fluorescence spectroscopy20,21, fluorescence anisotropy (FA)22,23, single-molecule fluorescence imaging24,25, and Bio-layer interferometry (BLI)26 are also either imprecise or incompatible with aptamer-small molecule interactions. Other principal issues of these methods are low sensitivity, high sample consumption, immobilization, mass transport limitations on surfaces, and/or buffer restrictions. Only a few of these technologies provide integrated controls for aggregation and adsorption effects.
MST represents a powerful tool for scientists to overcome this limitation to study the interactions between aptamers and small molecules27-29, as well as other targets such as proteins30-33. The technology relies on the movement of molecules through temperature gradients. This directed movement, called "thermophoresis," depends on the size, charge, and hydration shell of the molecule34,35. The binding of a ligand to the molecule will directly alter at least one of these parameters, resulting in a changed thermophoretic mobility. Ligands with small sizes may not have considerable impact in terms of size change from the unbound to the bound state, but they can have dramatic effects on the hydration shell and/or charge. The changes in the thermophoretic movement of molecules after interactions with the binding partner enables the quantification of basic binding parameters2,7,34,36,37.
As depicted in Figure 1A, the MST device consists of an infrared laser focused onto the sample within the glass capillaries using the same optics as for fluorescence detection. The thermophoretic movement of proteins via the intrinsic fluorescence of tryptophans6 or of a fluorescently labeled interaction partner3,8 can be monitored while the laser establishes a temperature gradient (ΔT of 2-6 °C). The resulting temperature difference in space, ΔT, leads to the depletion or accumulation of molecules in the area of elevated temperature, which can be quantified by the Soret coefficient (ST):
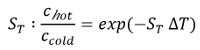
chot represents the concentration in the heated region, and ccold is the concentration in the initial cold region.
As shown in Figure 1B, a typical MST experiment results in an MST movement profile (time trace), consisting of different phases, which can be separated by their respective timescales. The initial fluorescence is measured in the first 5 s in absence of the temperature gradient to define the precise starting fluorescence and to check for photobleaching or photoenhancement. The Temperature Jump (T-Jump) represents the phase in which the fluorescence changes before thermophoretic movement. This initial decrease in fluorescence depends on heat-dependent changes of fluorophore quantum yield. The thermophoresis phase follows, in which the fluorescence decreases (or increases) due to the thermophoretic movement of the molecules until the steady-state distribution is reached. The reverse TJump and concomitant back diffusion of fluorescent molecules can be observed as indicated in Figure 1B after the laser is switched off. In order to access basic binding parameters, different molar ratios of the interaction partners are analyzed and compared. Typically, 16 different ratios are studied in one MST experiment, whereas the optical visible molecule is kept constant and is supplied with an increasing amount of the unlabeled ligand. The interaction between the two binding partners induces changes in the thermophoresis, and thus in the normalized fluorescence, Fnorm, which is calculated as following:
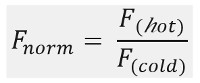
Fhot and Fcold represent averaged fluorescence intensities at defined time points of the MST traces. Binding affinities (Kd or EC50 values) can be calculated by curve fitting (Figure 1C).
Overall, MST is a powerful tool to study molecular interactions of any kind. This manuscript offers a protocol to characterize the challenging interaction between the small molecule adenosine triphosphate (ATP; 0.5 kDa) and the 25-nt short ssDNA aptamer DH25.42 (7.9 kDa). Over the course of the manuscript, the binding site of the aptamer on the ATP molecule is mapped down to the adenine group of the ATP.
In this study, MST was applied to characterize the binding site of the DH25.42 DNA aptamer18 on ATP. In contrast to other studies characterizing the interaction of ATP or ATP-mimicking small molecules with proteins randomly labeled with one or more fluorophores38-40, this study includes a labeled version of the 7.9 kDa ssDNA aptamer with one Cy5 molecule on the 5´ end. Different ATP derivatives and related molecules, all differing from ATP in various positions, were used to map the binding site on the ATP molecule. Dilution series (in 16 steps, reducing the ligand concentration by 50% each) of the different ligands were prepared and mixed with a constant amount of Cy5-labeled aptamer. Samples were analyzed in standard capillaries at 25% LED and 20% laser power. Movement profiles (MST time traces) were recorded and fluorescence units, derived from the T-Jump phase, were plotted versus the ligand concentration (compare Figure 1C). Curve fitting was performed applying the Hill equation, resulting in EC50 values. In 5 independent biological repeats (4 different operators, 2 different aptamer stocks, 2 different ATP stocks), the ATP-aptamer interaction shows a negative binding amplitude of 6 to 13 units and an average EC50 value of 52.3 ± 5.0 µM (Figure 2A). Error bars in the binding graphs and "±" presented in the affinity data represent the standard deviation of 5 biological repeats (5 independent measurements in a different capillary). Previously reported affinities for the interaction of the DH25.42 aptamer with [2,8,5'-3H]-adenosine and ATP agarose were comparable. The interaction of radiolabeled adenosine with the aptamer was quantified by a centrifugal filter assay, and an affinity (Kd) of 6 ± 3 µM was determined. Isocratic elution experiments with ATP immobilized to agarose resulted in an affinity (Kd) of 13 µM18,41.
For a better side-by-side comparison, the data can be normalized to the fraction of complexed molecules (fraction bound, FB) using the following equation:
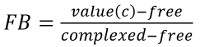
where value(c) is the MST value measured for the concentration c, free is the MST value for the unbound state (lowest concentration), and complexed is the MST value for the fully bound state (Figure 2B). This normalization is ideal to compare the results of different ligands in one graph, as shown in Figure 2C. The detected affinities of ADP (63.6 ± 5.9 µM in biological duplicates), AMP (91.6 ± 9.1 µM in biological duplicates), and SAM (44.4 ± 3.2 µM in biological duplicates), which differ from ATP in the number of phosphate groups, imply that this position has no or only minor influence on the binding behavior of the aptamer (Figure 2C and D). The OH group at the C2 carbon of the ribose of ATP could also be excluded from being the major binding site, as dATP was also bound by the aptamer with a slightly reduced affinity (64.4 ± 6.1 µM in biological duplicates). Changing the purine group of ATP to the pyrimidine group CTP resulted in non-binding of the aptamer, demonstrating the importance of this group for the interaction. The aptamer bound to adenine with an affinity of 141.7 ± 9.4 µM (in biological duplicates), showing that the binding site must be in this part of the ATP molecule. The purine molecules GTP and ATP differ in the green shaded area represented in Figure 2D, which represents the main binding site of the aptamer on ATP. Another study used non-quantitative elution experiments with different ATP derivatives to elute a radiolabeled aptamer from ATP agarose, which showed comparable results to this MST study18.
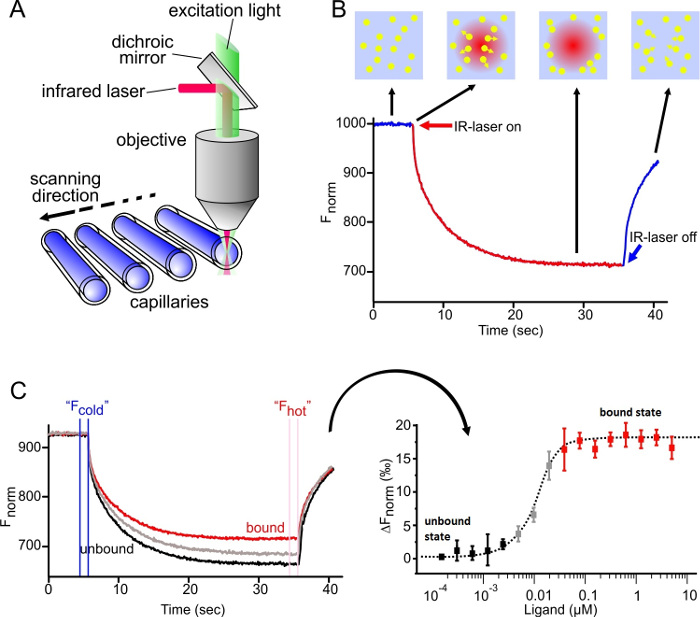
Figure 1: MicroScale Thermophoresis. (A) The technical setup of the MST technology is shown. The optics focus on the center of glass capillaries, thereby detecting the fluorescence signal of the optically visible molecule. An IR laser is utilized to establish a temperature gradient in the observation window of the optical system. Changes in fluorescence can be utilized to monitor the thermophoretic movement of the molecules in solution (B) MST time trace-movement profile of molecules in a temperature gradient. The initial fluorescence is measured for 5 sec while the laser is off. Switching on the laser generates a temperature gradient. After the immediate T-Jump phase, in which the fluorescent dye decreases its signal yield upon heat induction, the thermophoretic movement takes place and is observed for 30 sec. After the laser is turned off, the molecules diffuse back. (C) Results of a typical MST experiment: (Left) 16 capillaries containing the same concentration of fluorescent molecule and an increasing concentration of the unlabeled ligand; the MST time traces are recorded and normalized to their initial fluorescence. (Right) The normalized fluorescence; the difference between Fcold and Fhot is plotted against the concentration of the ligand. A curve fit of this data yields binding parameters, such as the binding affinity. Re-print with permission from Elsevier, Methods28; license number 3890230800113. Please click here to view a larger version of this figure.
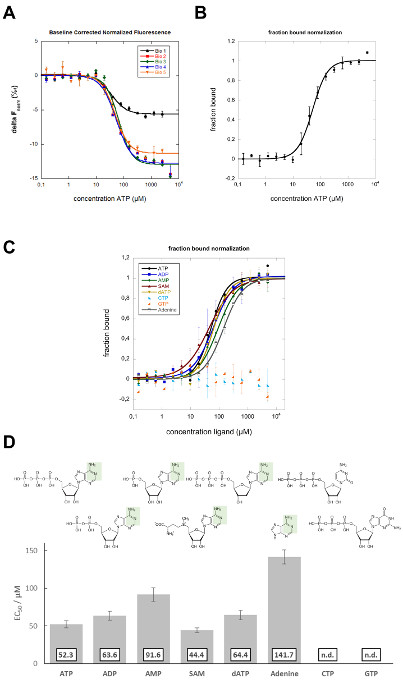
Figure 2: MST data analysis. (A) The baseline corrected normalized fluorescence ΔFnorm (‰), derived from the MST TJump signal, is plotted against the ATP concentration (in µM). The Hill equation (EC50) was applied for curve fitting. The error bars represent the standard deviation from five biological repeats. (B) Fraction-bound plot of the data shown in A. The respective data sets were normalized to the fraction bound, and the average of these normalized data is presented in the binding graph. The error bars indicate the standard deviation of 5 biological repeats. (C) The fraction-bound graph shows a quantitative comparison (Hill fit) of the different ligands to the aptamer. The error bars represent the standard deviation of two biological repeats. (D) Binding affinities (EC50) of different ligands to the aptamer. The green shaded area indicates the binding site of the aptamer on the adenine group of ATP. This figure is modified from Elsevier, Methods28; license number 3890230800113. Data sets from the previous study are reanalyzed and expanded within this study. Please click here to view a larger version of this figure.
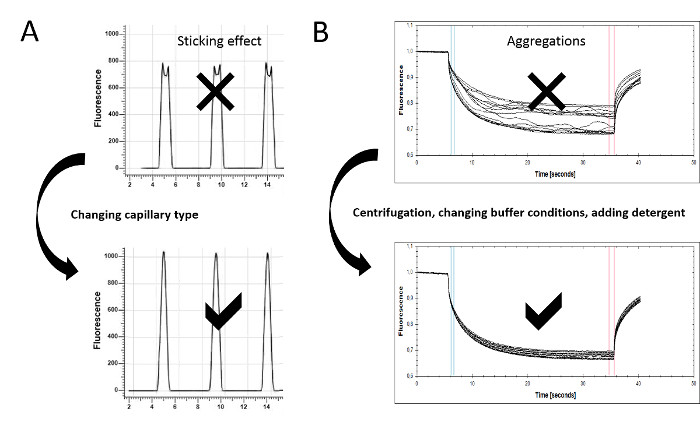
Figure 3: Assay optimization for MST experiments. (A) Protein adsorption and sticking effects can be detected in the capillary scan. Irregular peak shapes, such as flattened or U-shaped peaks, indicate the sticking of the sample material to the glass surface. Changing the capillary type (standard, premium, or hydrophobic) can prevent molecule adsorption, resulting in a regular peak shape. (B) The MST time traces also serve as a quality control, since aggregates can be detected there as bumps and spikes. The experimental conditions can be optimized by improving the solubility of the molecules (e.g., including detergents such as Pluronic F-127 or varying pH values or salt concentrations). Centrifugation may help to remove larger aggregates. To ensure optimal data quality, the MST time traces should resemble the given example. Please click here to view a larger version of this figure.
