The protocol described is summarized as a five-step workflow with images of the tissue at various stages of processing in Figure 1. The sample is first obtained through rapid sterile autopsy. During mechanical dissociation, the tissue is minced while removing blood vessels and meninges from the sample, and filtered through a 100 μm nylon mesh filter. Remaining tissue fragments are enzymatically dissociated in a warming oven. Next, debris is reduced through sucrose gradient centrifugation, isolating a distinct layer of myelin from the sample. In addition, ACK lysis visibly reduces the amount of red blood cells present in the sample. Finally, the cells are plated in serum-free media supplemented with growth factors.
Supplemental Figure 1 is an example of the tumor upon autopsy. The tumor grows as a diffuse infiltration of the pons and the adjoining regions of the brainstem. During sample preparations, small ~1 cm x 1 cm chunks should be cut and immediately placed into cold shipping media on ice.
Figure 2 shows various examples of how the samples can appear from the early stages of culturing to a durable culture. Initially plated and early cultures (A, B) often do not appear to contain many surviving cells. Furthermore, early cell clusters (C, D) often do not exhibit the degree of contrast typically associated with neurospheres. Notably, the duration of time in culture prior to the appearance of cell clusters can take weeks to a couple months. However, the initial passage of these cell clusters, combined with reverse filtering of cell clusters, can isolate healthy tumor cells that are able to form spheres (F). The filtrate (E) typically contains leftover debris; however, we recommend plating and maintaining the filtrate and monitoring for the development of cell clusters. These patient-derived samples can then be maintained in culture for multiple passages (G, H).
Figure 3 demonstrates the ability of these cells to be xenografted into mice. In each of these cases, the DIPG cells were transfected with a GFP-Luciferase reporter to monitor the development of the tumors through bioluminescence (A). The tumors can also be examined histologically, for example to observe tumor engraftment (B) or expression of specific markers (C). These in vivo models, combined with in vitro assays can be used to assess the effects of various drugs or gene manipulations (e.g.,8,9,10,11,14).
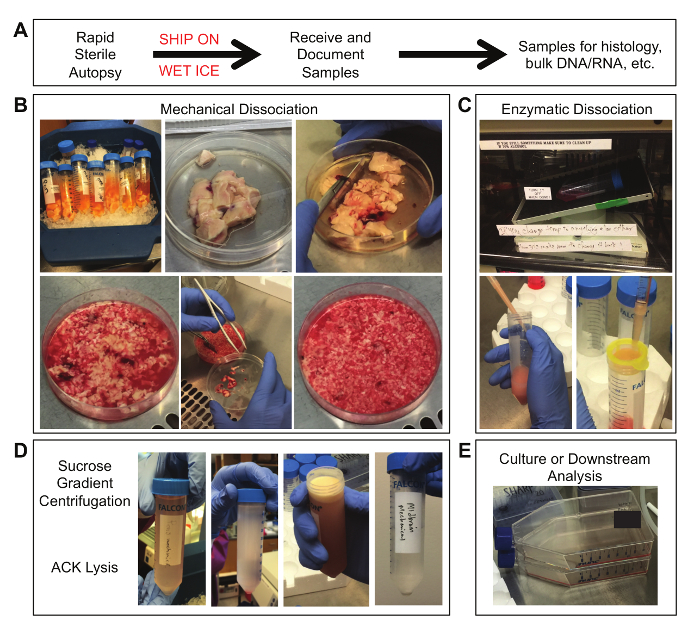
Figure 1. Tissue Samples at Various Stages of Processing. (A) Obtain the sample through sterile autopsy procedures and overnight shipping on wet ice. (B) From left to right: (row 1) tubes containing tumor tissue in shipping media; fresh tissue in a 100 mm x 20 mm cell culture dish; initial mincing of tissue; (row 2) partially minced tissue; removal of meninges and blood vessels; final size of minced tissue pieces. (C) From left to right: (row 1) tissue incubated on a rotating table in a 37 °C oven; (row 2) trituration of tissue through a 1000 μL pipet tip attached to a 10 mL pipet; filtering of dissociated tissue through a 100 μm nylon mesh filter. (D) From left to right: dissociated tissue suspended in 0.9 M sucrose solution prior to centrifugation; dissociated tissue separated across a sucrose gradient after centrifugation; a visible layer of myelin debris on top of the sucrose gradient; a final cell pellet after ACK lysis and wash. (E) The final sample can be placed into culture or used in other downstream analyses. Please click here to view a larger version of this figure.
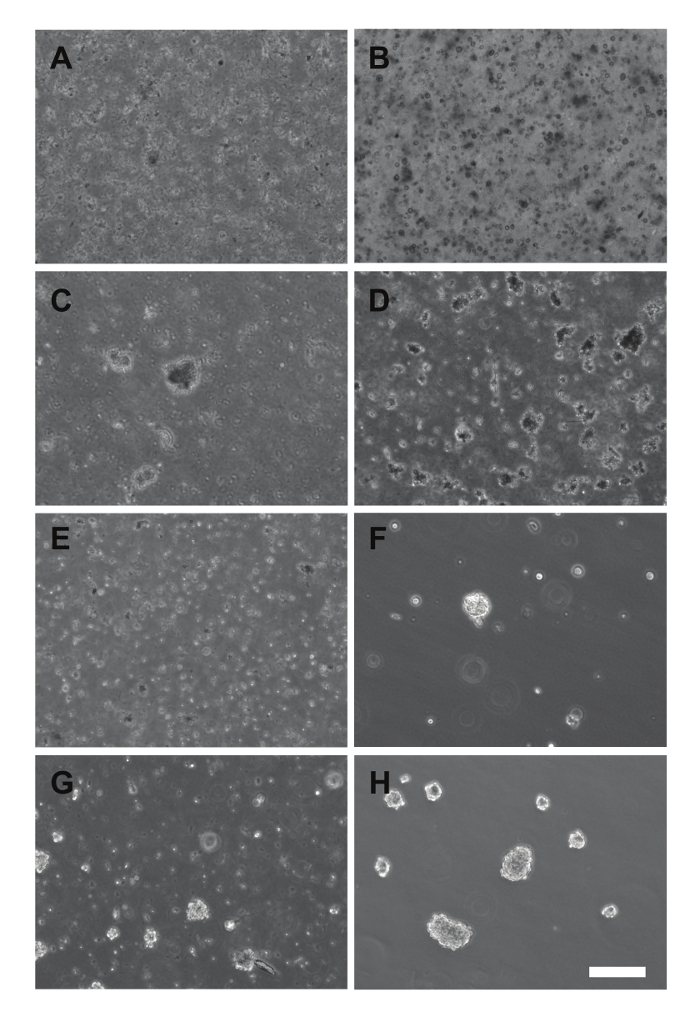
Figure 2. Images of Primary Cultures and Early Passage Cells. (A – B) Cultures plated immediately after dissociation (SU-DIPG-XXX, A), or that have not grown neurospheres yet (SU-DIPG-XXIX, B). (C – D) Early appearance of neurospheres in primary cultures of SU-DIPG-XXVIII (C) and SU-DIPG-XXIX (D). (E-F) First passage of the primary culture from D using a 100 μm nylon filter, with the filtrate (E) and reverse filtrate (F) plated. (G – H) Mature neurospheres from the first passage of SU-DIPG-XXVII (G) and the third passage of SU-DIPG-XXV (H) growing in culture. Scale bar = 200 μm. Please click here to view a larger version of this figure.
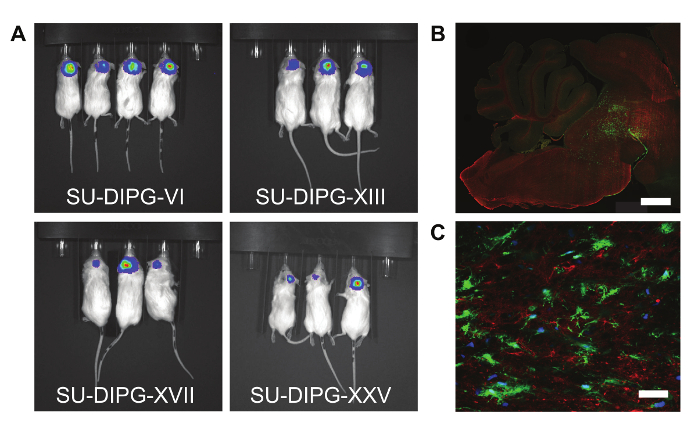
Figure 3. Patient-derived Cell Cultures Can Engraft and Form Tumors in Mice. (A) Bioluminescence images of four different patient-derived cell cultures transfected with a GFP-Luciferase reporter. (B) Sagittal section from a mouse xenograft, showing tumor cells engrafted in the mouse pons. Green: GFP, Red: Myelin basic protein. Scale bar = 1 mm. (C) Example immunofluorescence image showing GFP tumor cells engrafted in a mouse brain. Blue: DAPI, Green: GFP, Red: IGF2R. Scale bar = 40 μm. Please click here to view a larger version of this figure.
Supplemental Figure 1. Autopsy Sample of a Pontine Tumor. Immediate post-mortem appearance of a diffuse intrinsic pontine glioma. The tumor appears as a large, myelin-rich mass on the ventral surface of the pons. Please click here to download this image.
