This protocol describes an experimental framework for fluorescence imaging of living or fixed and stained Tribolium embryos with LSFM. Due to the low levels of photo-bleaching and photo-toxicity, a direct consequence of its optical sectioning capability, LSFM is particularly well suited for long-term live imaging.
The novel AGOC{ATub'H2B-mEmerald} #1 transgenic line expresses a histone2B-mEmerald fusion protein under control of the alpha-tubulin 1 promoter74. It shows an enhancer trap-like expression pattern51, since the fluorescence signal is mainly obtained from the serosa, the yolk sac and several neuronal cell clusters. Using this line, 66 hr of embryonic development at room temperature (23±1 °C) were recorded, covering germband retraction and dorsal closure (Supplementary Movie 1). This line can be used to visualize neuronal clusters within the head appendages (Figure 4A) and the dynamics of serosa migration during dorsal closure (Figure 4B). The novel AGOC{ATub'H2B-mEmerald} #4 line carries the same transgene as the #4 line, but at a different genomic location. It does not show an obvious enhancer trap pattern, but the fluorescence signal is spatiotemporally ubiquitously detectable as described previously for this promoter74. This line was imaged at the transition from gastrulation to germband elongation to demonstrate the optical sectioning capability at two depth levels (Figure 4C). Another feature, especially for live imaging, is the acquisition of z-stacks along multiple directions via sample rotation, which is standard for LSFM setups used in developmental biology. For example, in the Glia-blue58 line, sample rotation allows the observation of glia cell reorganization along and around the ventral nerve cord as well as the proliferation dynamics in the left head lobe, which can only be seen properly from the dorsal site, in the same embryo (Figure 4D, Supplementary Movie 2). The live imaging datasets associated with this study are provided as a downloadable resource, meta and access information are found in Supplementary Table 1.
Since the choice of transgenic lines for live imaging is still limited, small fluorescent dyes can be used to specifically label intracellular structures. SYTOX Green binds to cell nuclei and can be visualized in the green channel, while YOYO-1 and BOBO-3 Iodide bind to the nuclear envelope and can be visualized in the green or red channel, respectively (Figure 5A). These dyes can be used to highlight certain embryonic structures, such as the serosa scar (Figure 5B, first column), the posterior ventral serosa cells (Figure 5B, second column) or the serosa-amnion-germband tissue tri-layer that emerges during serosa window closure (Figure 5B, third to fifth column). Dual-color staining allows the visualization of two intracellular structures in the same embryo, for example the nuclear envelope together with the actin cytoskeleton, which can be stained with Alexa Fluor-conjugated Phalloidin dyes. For example, YOYO-1 Iodide can be combined with Phalloidin 546 (Figure 5C), while BOBO-3 can be combined with Phalloidin 488 (Figure 5D).
It is also convenient to fix and stain transgenic embryos that already express a certain fluorescent protein, since the fixation procedure quenches the intrinsic fluorescence signal only marginally. For example, embryos from the AGOC{ATub'H2B-mEmerald} #4 transgenic line, in which the nuclei are detected in the green channel, can be stained with Phalloidin 546 so that the actin cytoskeleton becomes visible in the red channel (Figure 6).
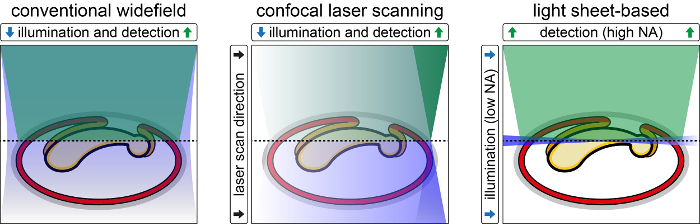
Figure 1: Comparison of Conventional Widefield, Confocal Laser Scanning and Light Sheet-based Fluorescence Microscopy. Conventional widefield epi-fluorescence microscopy uses xenon arc/mercury-vapor lamps with appropriate filters or high-power light emitting diodes as light sources. For each acquired two-dimensional image, the whole specimen is illuminated with a non-diffraction-limited light cone. In confocal laser scanning fluorescence microscopy, a diffraction-limited Gaussian laser beam is scanned through the specimen and fluorescence is detected incoherently point-by-point. Similarly to conventional widefield epi-fluorescence microscopy, the whole specimen is illuminated for each acquired two-dimensional image. In light sheet-based fluorescence microscopy, a diffraction-limited Gaussian laser beam illuminates the specimen perpendicularly to the detection axis. Fluorescence is detected either plane-by-plane or line-by-line. During the acquisition of a two-dimensional image, only a small volume centered on the focal plane of the detection objective experiences the effects of the excitation light. The remaining volume of the specimen is not illuminated, does not contribute to out-of-focus blur, does not suffer from photo-toxic effects and does not lose fluorophores due to photo-bleaching. Please click here to view a larger version of this figure.
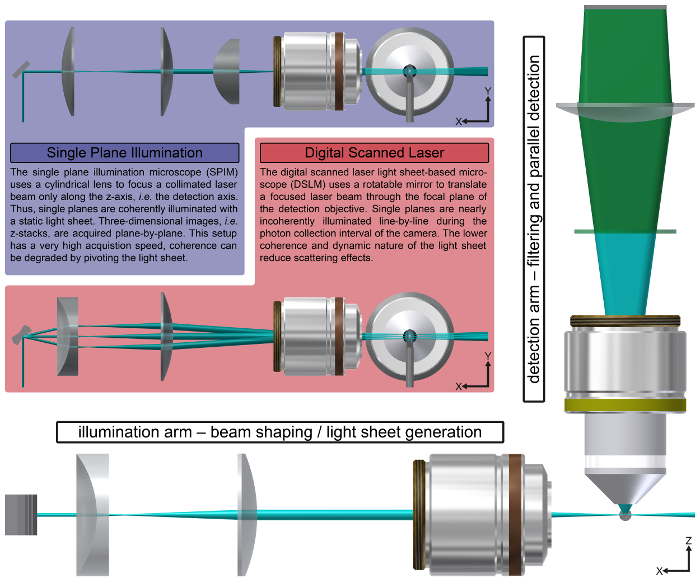
Figure 2: The Principle of Light Sheet-based Fluorescence Microscopy. In LSFM, illumination and detection are split into at least two optical paths. At least one illumination arm is used to generate the light sheet (cyan), while at least one detection arm is equipped with an appropriate filter and a camera for the parallel detection of the fluorescence signal (green). LSFM has two canonic implementations, the single (or selective) plane illumination microscope (blue background) and the digital scanned laser light sheet microscope (red background). By moving the specimen and the light sheet relative to each other, three-dimensional images are acquired. Information along multiple directions is collected by rotating the specimen around the y-axis, which is preferentially oriented along gravity. Please click here to view a larger version of this figure.
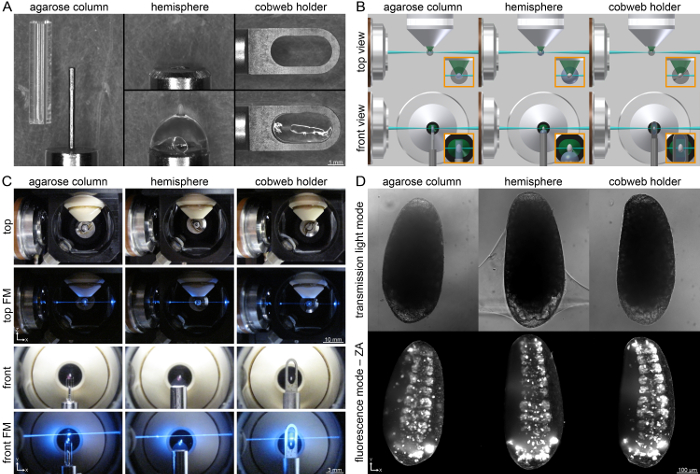
Figure 3: Three Different Mounting Techniques used in the Recording of the Embryonic Development of Tribolium with Light Sheet-based Fluorescence Microscopy. (A) The agarose column refers to a steel cylinder with a small pin that pushes the agarose column, in which the embryo is embedded, out of a glass capillary. The agarose hemisphere is mounted on a steel pipe. The embryo is attached to the pole of the hemisphere with a small amount of agarose. The cobweb holder is a sheet of metal with a slotted hole mounted on top of the steel cylinder. The embryo is glued to a thin agarose film that spans the slotted hole. (B) Schemes of the three mounting techniques inside a light sheet microscope. (C) The three mounting techniques applied within a DSLM. (D) Exemplary images from embryos of the Glia-blue transgenic line recorded with the three mounting techniques. The embryos, which are at the onset of germband retraction, are shown with their posterior end orientated towards top to match the transmission light images. FM, fluorescence mode; ZA, z maximum projection with intensity adjustment. Please click here to view a larger version of this figure.
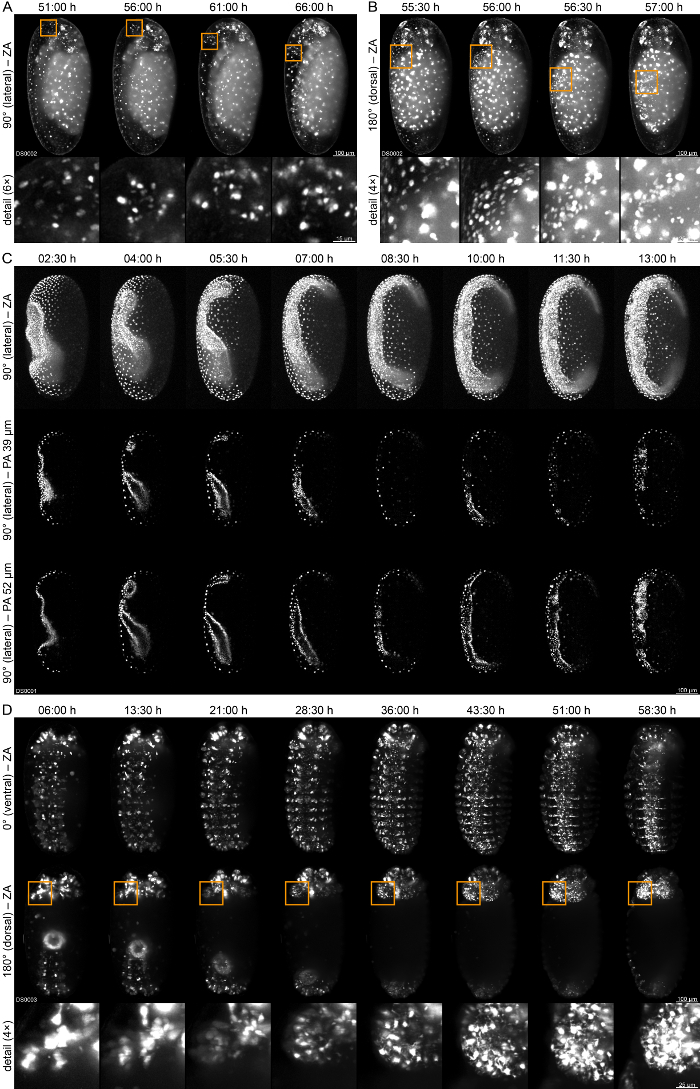
Figure 4: Live Imaging of Tribolium Embryos. (A) An embryo of the AGOC{ATub'H2B-mEmerald} #1 line at the transition from germband retraction to dorsal closure. This line exhibits an enhancer trap expression pattern. The fluorescence signal is mainly found in the serosa, the yolk sac and a few neuronal cell clusters. The embryo is shown over a period of 15 h with an interval of 5 h. The detail images show cell clusters in the head appendages. Initially, the clusters are still covered by serosal cells (first column), upon serosa rupture, the cells follow along with the turning movement of the head. (B) Same embryo shown during dorsal closure for 1:30 h with an interval of 0:30 h. The detail images show the migration of the ruptured serosa over the yolk sac. (C) Top row: An embryo of the AGOC{ATub'H2B-mEmerald} #4 line at the transition from gastrulation to germband elongation. Middle and lower row: Single planes are shown in two depths over a period of 10:30 h with an interval of 1:30 h. (D) An embryo of the Glia-blue line during germband retraction shown over a period of 50:30 h with an interval of 07:30 h. The detail images show the morphogenetic reorganization of the glial cells in the left head lobe. ZA, z maximum projection with intensity adjustment; PA single plane with intensity adjustment. Please click here to view a larger version of this figure.
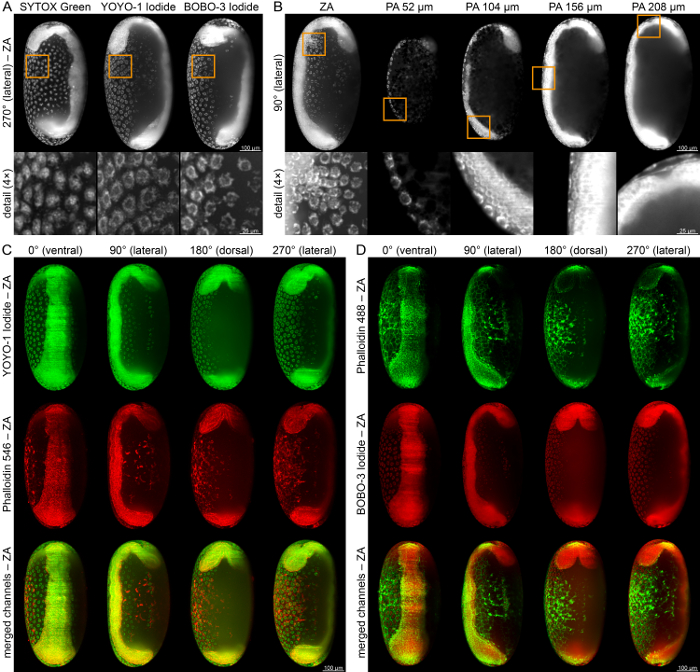
Figure 5: Fixation, Staining, and Iimaging of WT Embryos during Germband Elongation. (A) Embryos stained with either SYTOX Green, i.e. a fuzzy nuclear stain, or YOYO-1 Iodide or BOBO-3 Iodide, i.e. two nuclear envelope stains. (B) YOYO-1 Iodide-stained embryo. Details show the serosa scar (first column), the nuclear envelope of the large posterior-ventral serosa cells (second column) or the three-layered tissue organization of the serosa, the amnion and the actual embryonic tissue (third to fifth column). (C) Dual-color staining with YOYO-1 Iodide and Phalloidin 546. (D) Dual-color staining with Phalloidin 488 and BOBO-3 Iodide. ZA, z maximum projection with intensity adjustment. Please click here to view a larger version of this figure.
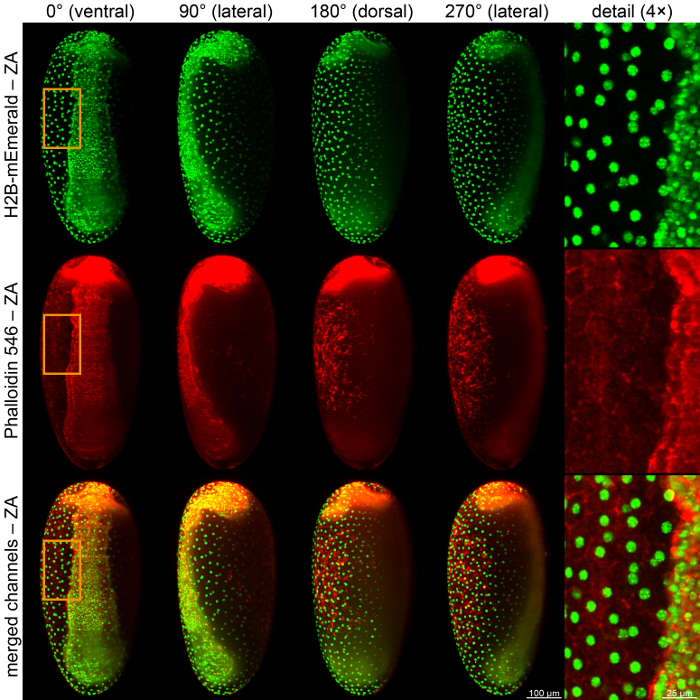
Figure 6: Images of Fixed and Stained Transgenic Embryos from the AGOC{ATub'H2B-mEmerald} #1 line. This transgenic line expresses mEmerald-labeled histone2B (H2B) under control of the alpha-tubulin 1 promoter, which marks the nuclei/chromatin ubiquitously throughout the whole embryonic development. The embryo was further stained with Alexa Fluor 546 Phalloidin, which labels the actin cytoskeleton/f-actin. ZA, z maximum projection with intensity adjustment. Please click here to view a larger version of this figure.
| criterion | fluorescence microscope type | ||
| conventional wide field | confocal laser scanning | light sheet-based | |
| objective lens setup | one objective lens, medium-high NA | one objective lens, high NA | two perpendicularly arranged objective lenses, illumination: low NA, detection: high NA |
| illumination light source | white light source and appropriate filters (e.g. mercury short-arc lamps) or LED-based with clean-up filters |
laser with clean-up filter (e.g. diode or DPPS lasers) |
laser with clean-up filter (e.g. diode or DPPS lasers) |
| illumination per two-dimensional image | whole sample | whole sample (typically two-dimensional Gaussian beam scan) |
only focal plane (DSLM: one-dimensional Gaussian beam scan) |
| detection | parallel (typically CCD or sCMOS camera) |
sequential (photomultiplier tube) |
parallel (typically CCD or sCMOS camera) |
| imaging speed | fast (milliseconds per image) | slow (seconds per image) | fast (milliseconds per image) |
| out-of-focus fluorescence | no discrimination | blocked nearly completely by the pinhole (rejection efficiency depends on diameter) |
nearly non-existent (only due to scattering) |
| spatial dimensions | 2 (x / y) |
3 (x / y / z) |
3 (x / y / z) |
| further degrees of freedom | 2 (time, fluorescence channel) |
2 (time, fluorescence channel) |
3 (time, fluorescence channel, direction) |
| lateral resolution | r (0.6 λ / NA) |
0.66 r (0.4 λ / NA) |
r (0.6 λ / NA) |
| optical sectioning capability | No | Yes (discrimination in the detection pathway) |
Yes (discrimination in the illumination pathway75) |
| availability and price | predominantly commercial, low cost | predominantly commercial, expensive | commercial, expensive custom-built, moderate54,59,60,75 |
| live imaging publications covering Tribolium embryos | six44,76,77,78,79,80 | seven23,77,79,81,82,81 (spinning disk confocal)83,84,85 |
four23,57,65,86 |
Table 1 – Characteristics of Fluorescence Microscopy Techniques used in Tribolium live Imaging.
| dye | staining | dilution |
| SYTOX Green | nuclei (fuzzy) | 1:10,000 (1:100 in ddH2O and 1:100 in 1% (w/v) BSA in PBS) |
| YOYO-1 Iodide | nuclear envelope | 1:10,000 (1:100 in ddH2O and 1:100 in 1% (w/v) BSA in PBS) |
| BOBO-3 Iodide | nuclear envelope | 1:10,000 (1:100 in ddH2O and 1:100 in 1% (w/v) BSA in PBS) |
| Alexa Fluor 488 Phalloidin | actin cytoskeleton | 1:100 (in 1% (w/v) BSA in PBS) |
| Alexa Fluor 546 Phalloidin | actin cytoskeleton | 1:100 (in 1% (w/v) BSA in PBS) |
Table 2 – Staining Solutions.
| agarose column (option 1) |
hemisphere (option 2) |
cobweb holder (option 3) |
|
| rationale | embryo is embedded in an agarose column that is pushed out of a capillary | posterior end of embryo is glued to pole of an agarose hemisphere | embryo is glued to thin agarose film spanning a slotted hole |
| sample holder | steel cylinder with pin | steel pipe | steel cylinder with slotted hole |
| directions (orientations) | unlimited (any) | unlimited (any) | four (0°, 90°, 180°, 270°) |
| required skill level | moderate | high | low |
| agarose around the embryo | high | low | moderate |
| number of embryos per session | up to three | one | up to six |
| embryo retrieval | tricky | easy | easy |
| disposable equipment | glass capillaries | – | – |
| recommended for | short-term live imaging, stained embryos | long-term live imaging | long-term live imaging, stained embryos |
| references | three23,87,88 | two57,65 | this publication |
Table 3 – Mounting Methods Advantages and Disadvantages.
| aberrational phenotype induction | rationale | step | reference (methodological) |
| embryonic RNA interference | double-stranded RNA is injected into embryos during the syncytial blastoderm stage to knock-down one or more specific genes | inject embryos after Step 4.8. | two33,81 |
| parental RNA interference | double-stranded RNA is injected laterally into female pupae between the abdominal segments 3 and 4, which leads to gene knock-down in the progeny | take the pupae from Step 1.5. | two36,37 |
| recessive embryonic lethal mutant lines | egg-laying cultures that carry a recessive embryonic lethal mutation heterozygous yield about 25% homozygote mutant progeny | cross mutant line into imaging line during Step 1.5. | three39,51,89 |
| application of extrinsic factors | certain bioactive or toxic factors are extrinsically applied to the embryos by adding them to the imaging buffer | add factor to imaging buffer during Step 2.2. | two83,85 |
Table 4 – LSFM Live Imaging of Aberrational Phenotypes
Supplementary Table 1 – Metadata and Parameter for the Long-term Live-imaging Datasets DS0001-0003. Please click here to download the file.
Supplementary Movie 1 – Live Imaging of a Tribolium Embryo from the AGOC{ATub'H2B-mEmerald} #1 Transgenic Line.
This line exhibits an enhancer trap expression pattern. The fluorescence signal is primarily found in the serosa, the yolk sac and a subset of neuronal cells. The embryo is shown along four orientations from 00:00 h to 66:00 h with an interval of 00:30 h between the time points. The movie starts at the beginning of germband retraction and ends once dorsal closure is completed. Frame rate is five frames per second. ZA, z maximum projection with intensity adjustment. Please click here to download the file.
Supplementary Movie 2 – Live Imaging of a Tribolium Embryo from the Glia-blue Transgenic Line.
The embryo is shown along four orientations from 00:00 h to 66:00 h with an interval of 00:30 h between the time points. The movie starts at the beginning of germband retraction and ends once dorsal closure is completed. Frame rate is five frames per second. ZA, z maximum projection with intensity adjustment. Please click here to download the file.
