Cloning and expression of different constructs of IKK1/α
Full length human IKK1/α was cloned into the baculovirus expression vector pFastBacHTa within its EcoRI and NotI restriction sites to obtain an N-terminal hexa-Histidine tagged IKK1. The tag could be removed by TEV protease digestion. Since full length IKK1/α contains flexible regions on both ends, and flexible regions usually render a protein difficult to crystallize, we cloned various truncated fragments of IKK1/α within the abovementioned sites of pFastBacHTa vector. Various truncation mutants of IKK1/α were generated with both wild-type (wt) and S176E,180E (EE) backbones. IKK1/α EE refers to the mimetic of the constitutively active form of the phosphorylated kinase. Recombinant baculovirus production and protein expression were performed using a previously published protocol with minor modifications43 (Figure 1).
Difficulty in IKK1/α crystallization
Since human IKK2/β and IKK1 are homologous, and we could crystallize different versions of human IKK2/β under several different conditions, we expected IKK1/α to crystallize under similar conditions using similar strategies. However, after extensive trials with several different IKK1 variants, we obtained crystals with only one truncated construct (IKK1 10-667 EE) (Figure 2), and that also only in the presence of the IKK inhibitor XII44 that displayed suitable X-ray diffraction properties.
Structure solution
IKK1/α crystals suffered from numerous growth problems, and the data often displayed very high mosaicity. Weak crystal packing associated with large solvent content and the dynamic nature of the IKK1 monomer/dimer are the likely reason behind this. To circumvent these problems, painstaking efforts were taken to obtain the best possible crystals, and the cryo-preservation procedure was performed with utmost care under various conditions and with various cryo-preservative buffers. The data was also collected with ultimate precision to minimize beam damage. Since the crystals were large (largest dimension often exceeding 400 microns), different areas of the same crystals were exposed to the X-ray beam to collect multiple datasets. This enabled different datasets to be scaled, and to obtain a reasonably complete dataset with higher redundancy and minimized error. Soaking with heavy atoms, or heavy atom clusters (e.g., TaBr and TAMM), caused the crystals to diffract inadequately. Although we could locate the position of TaBr clusters in derivative data, the poor quality of the data in addition to the non-isomorphocity provided little, if any, phase information.
We processed datasets with HKL2000 using various available parameters. The diffraction pattern revealed that the crystal belongs to the space group P21 with unit cell dimensions, a = 174.51, b = 186.94, c = 275.83 Å and β = 98.84 degrees. We used mainly I/σ to estimate the cut-off and tested various cut-offs for the datasets. We obtained a merged data set of 4.5 Å resolution from 4 of 7 sets of diffraction data collected on one crystal. We decided to keep data until 4.5 Å from visual inspection of the final density map. It may be worthwhile to use CC1/2, since we can analytically estimate CC of the merged dataset against the true (usually unmeasurable) intensities using CC*45. Because of the large unit cell combined with the low symmetry of the space group, we anticipated the asymmetric unit to contain 12 to 24 molecules based on a solvent content of 70% to 40%.
The combined effect of low resolution X-ray data with weak intensities (i.e., significant errors in data) (Figure 3), large number (12) of IKK1 molecules in the asymmetric unit, and conformational variation of IKK1 monomeric/dimeric model compared to known IKK2 models, initially prevented us from obtaining a molecular replacement solution IKK1, and thus determining its structure.
IKK1/α and IKK2/β both contain a kinase domain (KD), a ubiquitin-like domain (ULD), and an a-helical scaffold dimerization domain (SDD). The structure of IKK1 revealed that it forms dimers similarly to IKK2 utilizing a nearly identical inter-subunit interface of the distal region of SDD. However, IKK1 dimer displayed a significantly different relative positioning of the N-terminal KD relative to SDD+ULD compared to that in known IKK2 dimers. Different IKK2 structures earlier indicated different intermonomer orientation within its dimers (Figure 4, panel A), so that the distances between the alpha carbons of P578 in the two KD in four different dimer models varied between 39 and 61 Å. Since the sequences of these two kinases are very similar and residues at the dimer interface are identical, we anticipated IKK1/α would form a similar dimer; however, since there are significant differences in residues of the KD-SDD interface (e.g., W424 and F111 in IKK2 are V and P respectively), we anticipated perhaps a yet different KD orientation in IKK1. Indeed, IKK1/α structure indicated that the related orientations of KD to SDD is unique, and it deviates from all known models of IKK2/β. In excess of a hundred dimer models were used as search models in programs PHASER, MOLREP, and CNS without any success, indicating the need for a rather accurate search model for the success of molecular replacement trials, especially with our low-resolution data. A monomer model has too little mass to pick up any solution in the weak diffraction data of the large asymmetric unit containing 12 monomers. Also, the inclusion or omission of water in the search model made no difference in molecular replacement searches, especially because of the weak diffraction data. The CC1/2 and CC* indicates that data up to 4.5 Å is quite precise. We used a conservative resolution cut-off of 4.5 Å based on I/σ of 1.5. The datasets with different resolution cut-offs did not show any stark difference during molecular replacement search operations, and final maps refined against these datasets did not show any distinct improvement in map features upon extending resolution cut-offs. The final build of the model is quite accurate, more than what could be built from the density alone, likely since the initial molecular replacement models were built from models derived with a high-resolution data, and we utilized the cryo-EM map/density to cross-check the features of the map, especially in regions where maps were unclear.
In hindsight, the obtainment of a useful search model was possible only because of the availability of the low resolution cryo-EM map, and a rather high accuracy model of IKK1 domains that could be generated based on high resolution IKK2 structure (Figure 4, panel B). We could dock the IKK1 domains in the cryo-EM map of IKK1 to obtain a reasonably close dimer model. The initial model indicated an orientation of KD rotated about 24 degrees relative to that of a IKK2 monomer, and N-terminal opening of 58 Å (between P578 of two KDs in a dimer). Our prior knowledge of different IKK2/β dimer structures (and their variation) enabled us to fine-tune the search model by changing the openings between 30-62 Å. Using one of the dimers (52 Å opening) as a search model, we located six dimers in the asymmetric unit using program MOLREP46. These six dimers are organized into two hexamers in the asymmetric unit, and the calculated solvent has a volume of 68%.
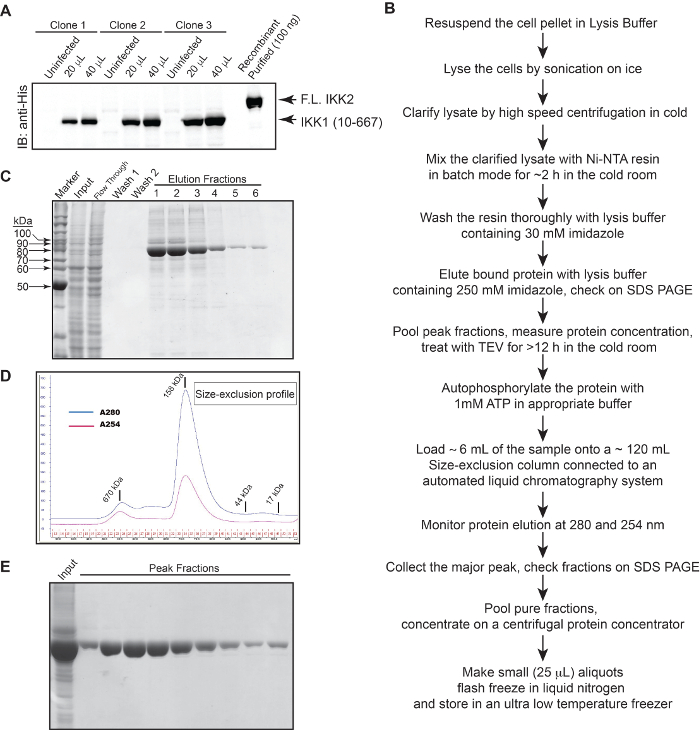
Figure 1: Purification of IKK1. (A) Expression of IKK1 (10-667) in insect cells. (B) A flow chart for purification of IKK1; (C) An SDS-PAGE gel showing purity of IKK1 after the Ni-NTA affinity chromatography step. (D) Size-exclusion profile indicating dimer of IKK1. (E) SDS-PAGE gel showing purity of IKK1 from the peak size-exclusion fractions. Please click here to view a larger version of this figure.
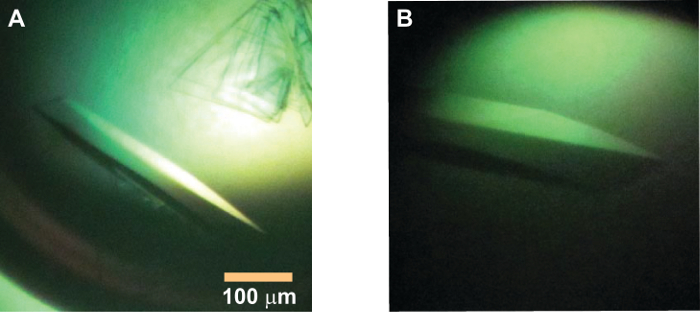
Figure 2: Morphology and size of IKK1 crystals. (A) A crystal of IKK1 construct 10-667; the crystallization drop also shows crystals of another morphology (thin plates) which do not diffract well. (B) Zoomed in view of the crystal that diffracted to beyond 5 Å. Please click here to view a larger version of this figure.
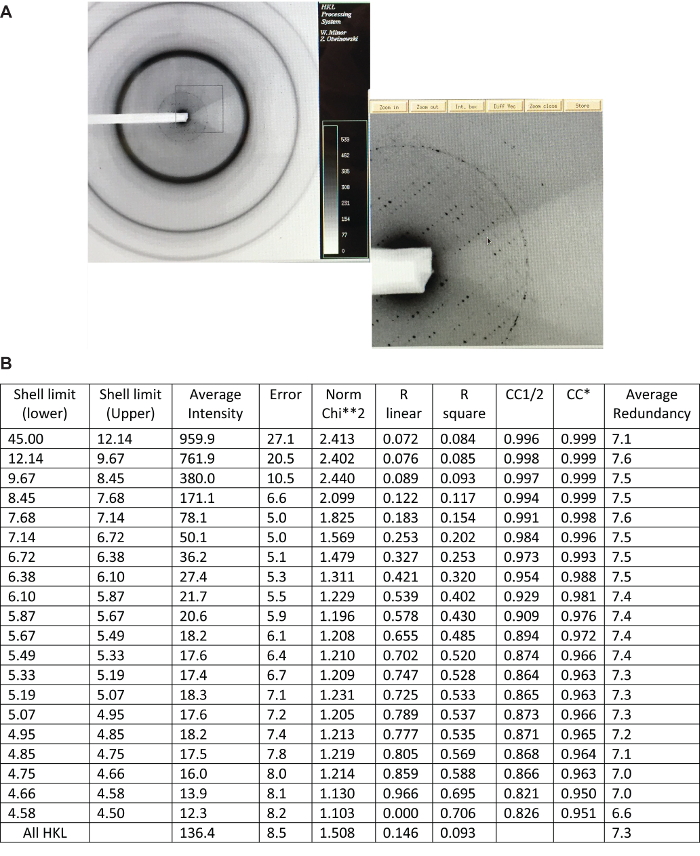
Figure 3:. Crystallographic data obtained from IKK1 crystals. (A) An X-ray diffraction profile of an IKK1 crystal indicating poor diffraction properties of typical IKK1 crystals. (B) HKL2000 scaling statistics of the merged dataset used for IKK1 structure determination, and redundancy of the data. R linear = SUM (ABS(I – <I>)) / SUM (I), R square = SUM ( (I – <I>) **2) / SUM (I **2), Chi**2 = SUM ( (I – <I>) ** 2) / (Error ** 2 * N / (N-1) ) ), CC1/2 = Correlation coefficient, CC* = Correlation coefficient of merged dataset against true intensities. Please click here to view a larger version of this figure.
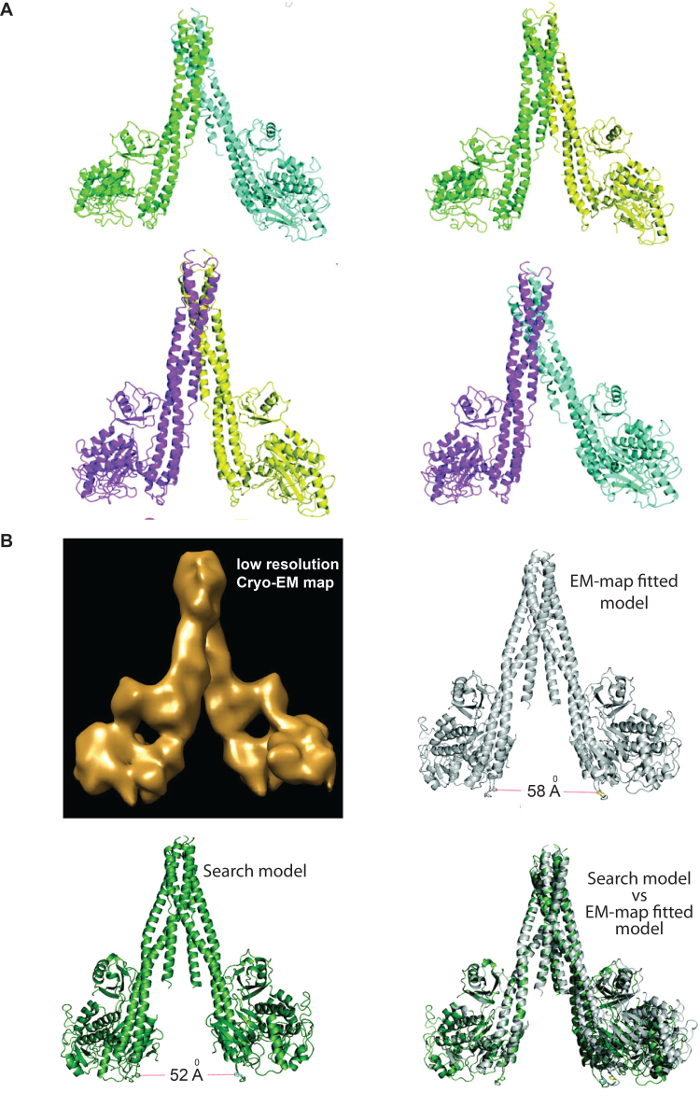
Figure 4: Preparation of search models. (A) Different models of IKK2 dimer indicating different orientations of the KD relative to SDD (giving rise to different separation between two KD); these models were tested initially to find a molecular replacement solution without any success. (B) Generation of IKK1 search models – Cryo-EM map of IKK1 dimer at 11 Å resolution; EM-map fitted initial IKK1 search model, individual domains are based on IKK2 model (4KIK); An IKK1 search model further fine-tweaked (appropriate KD orientation) based on various possibilities as judged from IKK2 models describe above in 4A; Superposition of EM-map fitted model to the search model that yielded the molecular replacement solution. Please click here to view a larger version of this figure.
